腓骨肌萎缩症1型患者肌电图及PMP22基因特点分析
2015-06-15潘晓丽潘志宏张楠楠高红
潘晓丽 潘志宏 张楠楠 高红
腓骨肌萎缩症1型患者肌电图及PMP22基因特点分析
潘晓丽 潘志宏 张楠楠 高红
目的 探讨和研究腓骨肌萎缩症1型(Charcot-Marie-Tooth disease 1,CMT1)患者肌电图和PMP22基因改变特点。方法 对43例CMT1患者进行常规神经传导速度和肌电图检查,应用PCR双酶切方法对其中33例CMT1患者及15名健康志愿者(对照组)检测17p11.2-12PMP22基因重复序列(即1760 bp片段)。33例CMT1患者依有无17p11.2-12PMP22基因特异性片段分为PMP22基因特异性片段阳性组与阴性组,比较两组患者神经传导改变有无差异。结果 43例患者均行肌电图检测,均表现为运动或感觉神经传导速度存在明显减慢(100%),感觉神经病变重于运动神经,下肢受累程度重于上肢;所检129块肌肉中,88块(68.2%)呈神经源性损害。经PMP22基因学检测的33例中20例(60.6%)检测出1760 bp片断,对照组均未检测到此片段。PMP22基因特异性片段阳性组感觉神经传导速度、运动神经传导速度及远端潜伏期与阴性组比较差异均无统计学意义(P>0.05)。结论 CMT1患者肌电图改变具有其特异性,结合PCR-双酶切法检测PMP22特异性基因重复序列可提高诊断CMT1的准确性及敏感性。
腓骨肌萎缩症;PCR-双酶切;PMP22基因;肌电图
腓骨肌萎缩症(Charcot-marie-tooth disease,CMT)是一组最常见的具有高度临床和遗传异质性的周围神经单基因遗传病,其发病率为4/10 000[1]。目前认为本病是一个综合征而不是独立性疾病,已将其归类在遗传性运动和感觉性神经病的范畴。CMT呈慢性进行性病程,致残率高,其病理变化主要发生于周围神经的远端部分、前根及后根,表现为对称性、节段性脱髓鞘改变及轴突变性。临床根据CMT的病理和电生理特点分为脱髓鞘型(CMT1)和轴突型(CMT2),其中CMT1约占70%。分子遗传学研究至少已发现40个致病基因位点,其中27个已被克隆[2-3]。本研究对43例CMT1患者肌电图和PMP22基因检测结果进行分析,期望对临床深入了解CMT有所帮助。
1 对象和方法
1.1 观察对象 2003-03-2013-03中国医科大学附属盛京医院就诊的CMT1患者43例,分别来自儿内科、儿骨科、发育儿科及神经内科门诊,共37个家系。其中男28例,女15例。发病年龄为4~19岁,中位数8.5岁(QL=3.5岁,QU=11.6岁),检测时病程为0.67~10年,中位数4.5年(QL=3.0年,QU=8.5年,)。无症状亲属(父母、祖父母、外祖父母等)52名。健康对照15名,男7名、女8名,年龄8~40岁,中位数13岁(QL=10岁,QU=32岁),均为除外周围神经病变、神经根性病变等神经系统疾病的健康志愿者。CMT诊断参考Dyck1993的标准[4]:(1)有慢性进行性双下肢远端无力及肌萎缩,可伴有双上肢受累;(2)查体有周围神经损害表现,跟腱反射消失,下肢感觉障碍;(3)肌电图示运动神经传导速度明显减低,正中神经传导速度<38 m/s;(4)排除吉兰-巴雷综合征及其他因素导致的周围神经病;(5)有明确CMT家族史支持诊断;(6)神经活检示有髓纤维数量减少,有髓鞘脱失,Schwann细胞节段性再生,绕轴突形成同心圆样的“洋葱球”结构,可以确诊。符合上述1~4条患者即可入选。本研究经中国医科大学医学伦理委员会批准,受试者本人或监护人知情并同意。
1.2 方法

1.2.2 PCR法检测PMP22基因:取外周血5 mL,2%(质量浓度)EDTA抗凝,常规提取DNA,DNA溶于10 mmol/L Tris-HCl,1 mmol/LEDTA溶液中(pH 7.5)保存。引物由invitrogen英骏生物技术有限公司合成。PMP22(peripheral myelin protein,PMP22)引物:远端为5′-GCTCAATTTTGGTGTTTAAATCAG-3′,近端为5′-ACTCTCTAGTTACCCCTTTATTT-3′。反应体系中DNA 5 μL,dd.H2O 292.6 μL,10X缓冲液55 μL,dNTPs 44 μL(2.5 mmol/L),TaqDNA多聚酶4.4 μL(5 U/μL),Pmp22-F 及Pmp22-P各2.2 μL。94℃预变性3 min后,进入循环。循环参数:94 ℃ 30 s,50℃ 60 s,72℃ 2 min,33个循环后72℃延伸10 min。取产物5 μL+EcoRⅠ(or NsiⅠ)12 U+缓冲液2 μL,37 ℃水浴酶切过夜,0.8%(体积分数)琼脂糖凝胶电泳,EB染色,存入10 Kodak成像分析系统。参照Marker相对分子质量标准,1760 bp条带为特异性酶切片段,有此片段诊断为基因重复。
将进行PMP22基因检测的患者按有无特异性片段分为PMP22基因特异性片段阳性组及阴性组两组,对两组患者感觉神经及运动神经传导速度进行比较。
1.3 统计学处理 应用SPSS16.0软件进行分析。神经传导速度用均数±标准差表示。PMP22基因特异性片段阴性组及阳性组患者感觉神经及运动神经传导速度比较采用独立样本t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 临床特点 本组患者28例(65.2%)为散发病例;15例(34.8%)有家族史,来自9个家系,其中有3例患者的家系1个,有2例患者的家系4个(其中2个家系有两例患者由于年龄较大,行动不便没有参加本研究),有1例患者的家系4个(其中1个家系的1例患者由于行动不便没有参加本研究)。患者均为慢性起病,表现为进行性双下肢或四肢对称性远端无力和肌萎缩,进展缓慢。症状局限于双下肢者29例(67.4%),呈跨阈步态,患者均有不同程度弓形足,其中10例曾被误诊为马蹄内翻足进行过矫形手术。前臂或手肌萎缩13例(30.2%)。“鹤样腿”13例(30.2%)均为病程大于5年的患者。腱反射减弱或消失29例(67.4%),深浅感觉减退20例(46.5%)。所有患者均无锥体束征,血清肌酸激酶、乳酸脱氢酶、谷草转氨酶、肌红蛋白等均在正常范围。
2.2 神经传导速度 结果见表1。43例患者均被检测到运动或感觉神经传导速度存在明显减慢,感觉神经病变重于运动神经,下肢受累程度重于上肢。即使是症状局限在下肢的患儿,上肢神经传导也均出现了明显的异常改变,总异常率100%。患者分别表现为不同神经传导速度明显减慢,其中感觉神经传导速度均低于正常下限的80%,运动神经传导速度均低于正常下限的75%。上、下肢运动神经传导速度均<38 m/s,远端潜伏期明显延长,均高于正常上限的150%,波幅降低。
2.3 肌电图 共检测43例患者129块肌肉,88块呈神经源性损害(68.2%),其中76块为远端肌肉,12块为近端肌肉;18块可疑神经源性损害(13.9%),其余23块正常(17.8%)。43例中37例(86.1%)存在神经源性损害,远端肌肉均呈神经源性损害,近端肌肉呈神经源性损害者12例。
2.4PMP22基因特异性片段检测 43例患者中有33例(包括来自8个有家族史的家系的患者13例)进行了PMP22基因学检测。20例(60.6%)检测出1760 bp片段,其中有6例父亲、9例母亲检测出同样的异常。对照组未检测到此片段(图1)。
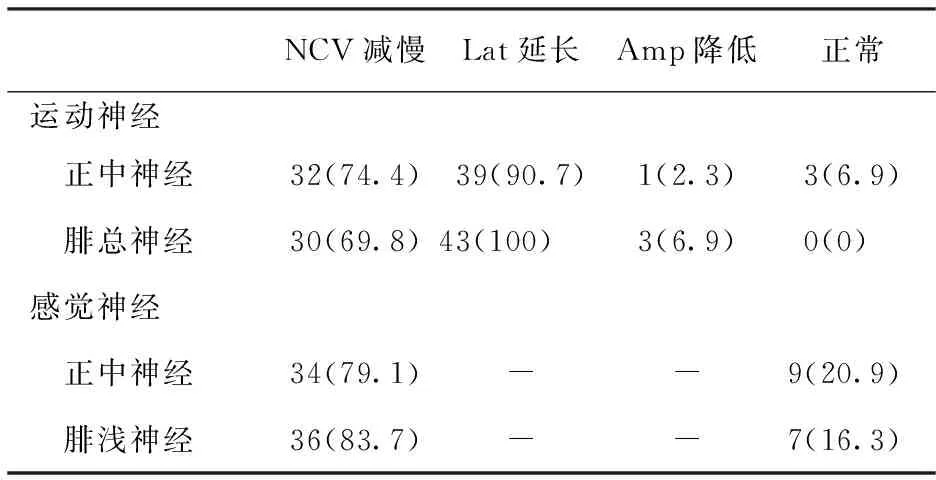
表1 43例CMT1患者神经传导检测结果 〔n=43,n(%)〕
注:NCV:神经传导速度;Lat:远端潜伏期;Amp:复合肌肉动作电位波幅

1、5、6、7为基因重复的患者;C为健康人(阴性对照);2、3为未检出重复的患者;M为DNA Marker DL2000
图1 CMT1患者及对照组PCR-双酶切分析结果
2.5 有、无PMP22基因特异性片段患者神经传导速度比较 结果见表2。两组患者不同感觉神经、不同运动神经传导速度下降程度及远端潜伏期延长程度等差异均无统计学意义(均P>0.05)。

表2 PMP22基因特异性片段阳性组与阴性组神经传导速度异常比较±s,%)
注:SCV:感觉神经传导速度; MCV:运动神经传导速度;Lat:远端潜伏期
3 讨论
目前的研究认为,CMT1为常染色体显性遗传,一般在10~20岁时发病, 起病隐袭, 国内报道的平均发病年龄为20~26岁[5], 本组病例发病年龄最早为4岁,中位数8.5岁,明显低于此前报道,可能与本研究大部分病例来源于儿科有关,所选患者本身年龄偏小,样本数量有限,因此尚需进一步研究。43例患者中只有15例有家族史,28例为散发病例。所有患者均符合CMT1的临床特征,临床上不难诊断。但是本研究中有10例CMT1患者,由于发病年龄较小并出现足部内翻畸形而误诊为马蹄内翻足进行过矫形手术,因病因未除,术后效果不佳。因此建议临床对马蹄内翻足患儿应常规进行术前肌电图检查,既可评价神经肌肉功能,亦可除外CMT1。CMT1患儿的肌电图改变具有明显特征,而且其不仅局限在下肢,上肢也会有明显异常,尤其是正中神经的运动神经传导速度均<38 m/s。
CMT1进展缓慢,肌电图可以显示慢性去神经支配和神经再生现象,表现为纤颤电位、正锐波和束颤电位,以及长时程、高波幅的运动单位电位并且募集相减少,神经传导速度表现为不同程度的减慢。本组患者所检测的129块肌肉中,88块呈神经源性损害(68.2%)。以远端肌肉病变较为严重;周围神经感觉及运动神经传导均有明显减慢,无论病程长短,运动神经传导速度均<38 m/s。之所以出现传导速度的减慢,主要是由于节段性的脱髓鞘,使得在有髓区域不能进行跳跃式的传导,其次大纤维选择性丧失,导致传导最快的纤维速度减慢。一些患者虽然临床症状局限于下肢,但上肢的神经传导异常已经存在,这表明在临床症状出现前已经有周围神经的脱髓鞘改变。肌电图是对神经肌肉功能评价最有效的检查手段,临床上应对那些起病缓慢、隐袭、临床表现不典型,患者不能提供遗传史的散发病例,进行四肢肌电图和神经传导速度检查,可有效地减少临床的漏诊和误诊,由于周围神经脱髓鞘的异常改变随病情进展,可在神经传导等电生理指标上得到准确的体现,因此肌电图检查可以评价患者的疾病进展程度。本研究进一步分析发现,PMP22基因特异性片段阳性组与阴性组神经传导速度异常特征性比较差异无统计学意义,提示肌电图在不同基因型CMT1患者的鉴别上无特殊价值。
目前研究发现70%以上相互无血缘关系的CMT1家系(独立家系,每个家系内有2例以上CMT1患者)及90%以上的散发患者多由17p11.2-12区(包含PMP22基因)的1.5 Mb的正向串联重复突变所致,两翼有长约30 kb的低拷贝重复序列称REPs。目前已证实CMT1的交换热点位于30 kb的CMT1A-REP上1.7 kb的区域,并且75%以上的CMT1患者交换断裂点位于该区域;对断裂点设计特定的引物进行扩增酶切获得特异性酶切片段即可诊断为基因重复异常[6-7]。PMP22在周围神经中有高度表达,其表达受轴突调节,其功能可能为维持髓鞘结构的完整性、调节细胞周期及作为黏附分子。PCR-双酶切法的原理:大多数CMT患者的重组断点位于远端CMT1-REP特有的EcoR Ⅰ位点和近端REP特有的Nsi Ⅰ位点之间。为区分PCR扩增,引物的选择分别为远端和近端REP所特有的序列,位于1.7 kb热点区的两侧。用EcoR Ⅰ和Nsi Ⅰ消化后可获得特异性融合片段1760 bp。这样来自CMT患者的标本且交换热点位于1.7 kb区域者可以产生特异性融合片段,而来自对照组的标本或重组发生于热点区外者,不应有此片段。本研究共检测33例患者,20例(60.6%)检测出1760 bp片段。其中13例有家族史的患者中均检测出此片段。健康对照组无检测到此片段者,总检出率低于文献报道[8-9]。本组患者均为CMT1,肌电图改变主要以周围神经传导速度减慢为主,但由于该病存在很大的遗传异质性,所以在遗传学改变上不尽相同,尚需进一步对本组患者进行其他相关基因的研究。
[1]Braathen GJ. Genetic epidemiology of Charcot-Marie-Tooth disease[J]. Acta Neurol Scand Suppl,2012,(193): iv-22.
[2]Saporta AS, Sottile SL, Miller LJ, et al. Charcot Marie Tooth (CMT) subtypes and genetic testing strategies[J]. Ann Neurol, 2011,69:22-33.
[3]Moszynska I, Kabzninska D, Sinkiewicz-Darol E. A newly identified Thr99fsX110 mutation in thePMP22 gene associated with an atypical phenotype of the hereditary neuropathy with liability to pressure palsies[J]. Acta Biochim Po, 2009,56(4):627-630.
[4]Dyck PJ, Chance P, Lebo R, et al. Hereditary motor and sensory neuropathy[M]. Philadephia: WB Saunders, 1993.1094-1132.
[5]梁银杏,葛辉,廖松洁, 等. 遗传性运动感觉性周围神经病Ⅰ型的临床及电生理特点[J].中国神经精神疾病杂志, 2010,36(4):217-219.
[6]Vital A, Latour P, Sole G, et al. A French family with Charcot-Marie-Tooth disease related to simultaneous heterozygous MFN2 and GDAP1 mutations[J]. Neuromuscul Disord, 2012, 22(8):735-741.
[7]Jones EA, Brewer MH, Srinivasan R, et al. Distal enhancers upstream of the Charcot-Marie-Tooth type 1A disease genePMP22[J]. Hum Mol Genet, 2012,21(7):1581-1591.
[8]Kim YH, Chung HK, Park KD, et al. Comparison between clinical disabilities and electrophysiological values in Charcot-Marie-Tooth 1A patients withPMP22 duplication[J]. J Clin Neurol, 2012,8(2):139-145.
[9]Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing[J]. J Neurol Neurosurg Psychiatry, 2012,83(7):706-710.
(本文编辑:邹晨双)
Electromyography andPMP22 gene analysis in patients with type 1 Charcot-Marie-Tooth disease
PANXiaoli*,PANZhihong,ZHANGNannan,GAOHong.
*NeuroelectrophysiologyLab,DepartmentofNeurology,ShengjingHospital,ChinaMedicalUniversity,ShenyangLiaoning110004,China
PAN Xiaoli,Email: panxl@sj-hospital.org
Objective To study the electromyography andPMP22 gene features in patients with type 1 Charcot-Marie-Tooth(CMT)disease. Methods Routine electromyography and nerve conduction were performed in 43 patients with CMT 1. Polymerase chain reaction(PCR) combined with restriction enzyme digestion was used to detectPMP22 gene duplication on chromosome 17p11.2-12 (1760 bp)in 33 CMT 1 patients and 15 healthy volunteers (the control group). According to the presence or absence of 17 p11.2-12PMP22 gene segments, 33 CMT 1 patients were divided into the positive group and the negative group. Parameters of nerve conduction were compared between two groups. Results All of the patients had the nerve conduction velocities slower or disappeared (43/43,100%). Sensory nerves and motor nerves were probably damaged at the same time, but the damages of the sensory nerves were more severe than those of the motor nerves, the damages of the lower limbs were more severe than those of the upper. Of 129 muscles tested, 88 (68.2%) showed neurogenic damages. 60.6%(20/33)of the patients were identified to have specific junction fragments(1760 bp). Duplication was not identified in the controls. There were no statistically significant difference of the parameters, such as sensory nerve velocity, motor nerve conduction velocity and distal latency between two groups (P>0.05). Conclusions Electromyography has its specificity in CMT 1 patients, in combination with double enzyme digestion method by PCR to detectPMP22 repetitive sequence specific genes can increase the accuracy and sensitivity in the diagnosis of CMT 1.
Charcot-Marie-Tooth disease;PCR-double enzyme digestion;PMP22 gene;electromyography
10.3969/j.issn.1006-2963.2015.02.002
110004中国医科大学附属盛京医院神经功能室(潘晓丽、张楠楠);110013沈阳市红十字会医院骨科(潘志宏);110004中国医科大学附属盛京医院 先天畸形实验室(高红)
潘晓丽,Email:panxl@sj-hospital.org
R746.4
A
1006-2963 (2015)02-0082-04
2014-01-09)
