新型喹唑啉酮类化合物合成工艺研究
2015-05-24赵蕾,赵雪梅,唐辉等
·论著·
新型喹唑啉酮类化合物合成工艺研究
赵 蕾1,2,赵雪梅1,唐 辉1,吴 娟3(1.山东大学附属省立医院,山东 济南 250021;2.山东大学齐鲁医院,山东 济南 250012;3.成都军区总医院,四川 成都 610083)
目的优化取代喹唑啉酮类化合物的合成工艺,重点考察温度、时间2个因素对关键中间体化合物合成的影响。方法平行试验比较温度、时间和缩合剂催化酯键的氨解反应,并对试验结果进行整体化分析;采用正交设计的方差分析考察反应物、温度及时间对收率的影响。结果和结论所得化合物的结构经过1H NMR、MS和13C NMR等方法确证,当反应温度为30℃,反应时间为24h,缩合剂(DCC)与反应物的投料比为1.5∶1时,酯键氨解反应的收率最高(65.41%),使得取代喹唑啉酮类化合物的合成路线更加符合工业生产的要求。体外抗真菌活性实验结果表明,所测定的先导化合物对5种临床致病菌都具有潜在的抗真菌活性,值得进一步研究。
喹唑啉酮类化合物;合成;正交设计;方差分析
喹唑啉酮类化合物在医药和化工领域是一类具有优良生物活性和生理活性的中间体化合物,该类化合物主要抑制表皮生长因子受体(EGFR)或其酪氨酸激酶(EGFR-TK)、血小板衍生生长因子受体(PDGFR)、神经生长因子受体(NGFR)和血管内皮生长因子受体(VEGFR)等受体的活性[1,2],广泛应用于抗真菌、抗肿瘤、抗病毒、抗疟疾等药物化学和有机化学合成[3,4]。由于喹唑啉酮类化合物具有特殊的结构与化学性质,及研究者对其先导化合物的多作用靶点的研究,对进一步探索针对不同原料与方法合成出更强生物活性的化合物具有重要意义[5]。在研究喹唑啉酮类化合物作用靶点和生物活性的同时,对该类化合物合成工艺新方法的探索也具有更深层次的意义[6,7]。
本实验参考相关文献[8-10],采用2-氨基-5-甲基-苯甲酸、丙酸酐和胺类化合物为原料,经多步反应生成相应的喹唑啉酮类抗真菌药物的衍生化合物。其中,反应的温度和时间为关键因素,从2个关键因素出发,对该路线的工艺进行优化,得到最优的工艺路线。
1 合成部分
2-乙基-6-甲基-3-戊基-3H-喹唑啉-4-酮的合成方法见图1。
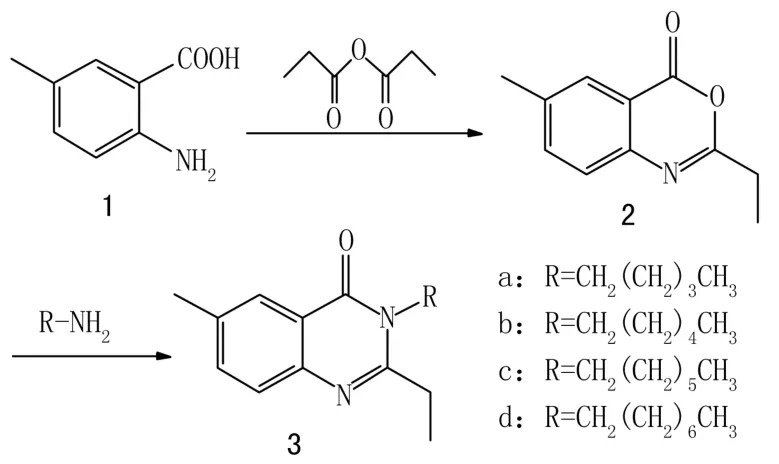
图1 2-乙基-6-甲基-3-戊基-3H-喹唑啉-4-酮的合成路线
2 实验部分
2.1 仪器和试剂 DF-101S型集热式恒温加热磁力搅拌器;JB-3型定时恒温磁力搅拌器;减压浓缩旋转蒸发仪;隔膜真空泵;电热恒温干燥箱;电子天平;玻璃仪器气流烘干箱;旋转蒸发仪;硅胶GF254板;所有试剂均为分析纯。
2.2 实验步骤
2.2.1 2-乙基-6-甲基-苯丙[1,3] 嗪-4-酮的合成[8] 取2-氨基-5-甲基-苯甲酸(6.0g,0.04mol)与丙酸酐40m l,加热搅拌回流5h,停止反应,放冷至室温后,于冰箱内(4℃)放置过夜,滤集析出的白色片状晶体,得粗品。用无水乙醇重结晶得淡黄色化合物2,4.66g,产率61.6%,mp 100.0~100.5℃(文献值:99.1~99.9℃)。
同法,用化合物2分别与正己胺、正庚胺、正辛胺反应,制备3b、3c、3d。
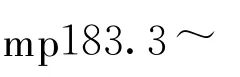
3 正交试验设计的方差分析
化合物2经酯键氨解反应得到化合物3,该反应过程涉及反应温度和反应时间2个因素。本试验以反应温度(A)、反应时间(B)为影响因素,做正交试验设计的方差分析,按照正交表的设计安排9次反应,以化合物3的收率作为判断指标。考察A、B两因素对总体工艺的影响[11]。研究因素水平列于表1。
根据试验结果(表2)和正交试验设计的方差分析(表3),判断影响反应结果中收率的因素,最优反应条件为:在30℃下反应24h,反应条件A2B2的产率最高。

表1 正交试验设计的影响因素水平
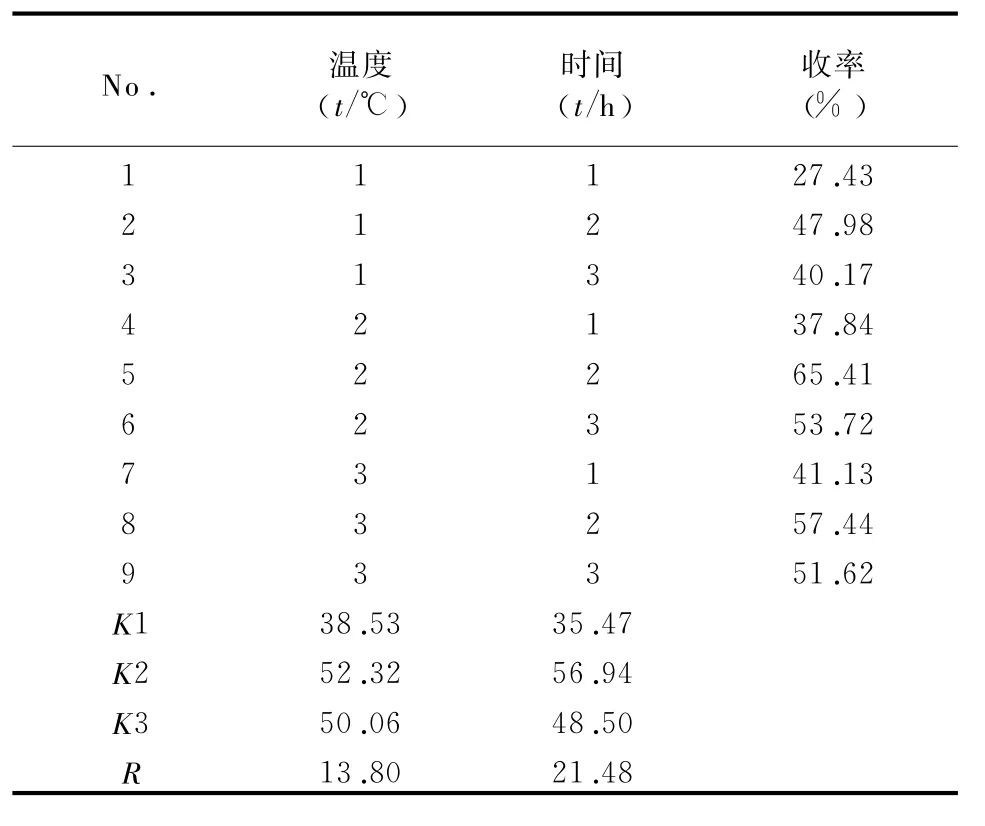
表2 正交设计试验结果
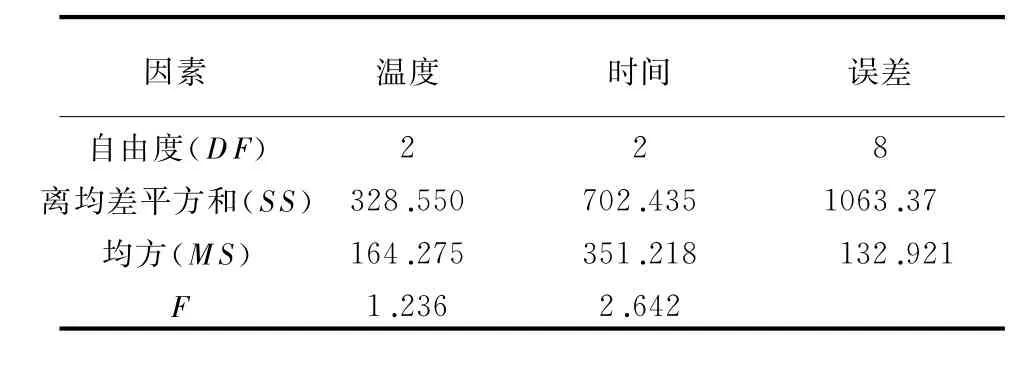
表3 正交试验设计方差分析
在本实验的反应条件中,缩合剂与反应物的投料比也是影响收率的一个重要因素,合适的投料比能够大幅提高反应收率。本实验选用DCC为缩合剂,在30℃下反应24h,以化合物3的收率作为判断指标,考察DCC对总体反应收率的影响(表4)。

表4 缩合剂DCC与反应物的投料比对反应收率的影响
由表4可以看出,当缩合剂DCC与反应物的投料比为1.5∶1时,基本达到最大收率(65.41%)。继续增大其原料摩尔比,由于产生较多的副产物,产物收率不升反降,后处理较为复杂,故缩合剂与反应物的投料比以1.5∶1较为适宜。
5 抗真菌活性的测定
参照美国国家临床试验标准化委员会(NCCLS)提出的药敏试验方案,采用微量液基稀释法,测定先导化合物对5种常见致病真菌的体外抗真菌活性[12,13]。
5.1 实验菌株 本实验测试菌株为5种常见的深部及浅表人体致病真菌,即白色念珠菌、新生隐球菌、烟曲霉菌、红毛癣菌、石膏样小孢子菌。
5.2 实验方法 采用RPM I 1640培养基,测定药物的最低抑菌浓度(M IC)。被测定的先导化合物分别用DMSO溶解配成,以氟康唑作为对照药物。用刚制作完成的RPM I1640培养基溶液将所要测定的先导化合物稀释至以下质量浓度:64、32、16、8、4、2、1、0.5、0.25、0.125μg/m l。分别接种于所要测定的实验菌株上,于37℃培养,观察白色念珠菌24h的生长情况;观察新生隐球菌72h的生长情况;其余菌株观察7d的生长情况。抗真菌活性见表5。
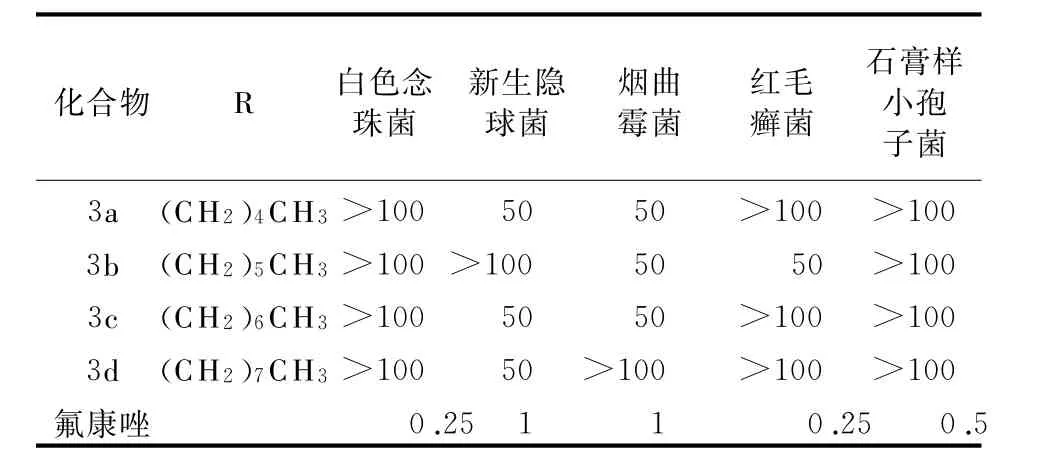
表5 先导化合物的体外抗真菌活性(ρB/μg·m l-1)
6 结果与讨论
本实验合成的4个化合物结构经过1H NMR、MS和13C NMR等方法确证,所有目标化合物均未见文献报道。其中,反应原料2-氨基-5-甲基-苯甲酸廉价易得,且处理方法较为简便,反应终产物杂质少、易分离纯化。根据反应的最终实验结果,提出最佳反应条件:当反应温度为30℃,反应时间为24h,缩合剂DCC与反应物的投料比为1.5∶1时,酯键氨解反应的收率最高,为65.41%。体外抗真菌活性实验结果表明,所测定的先导化合物对5种临床致病真菌均具有潜在的抗真菌活性,值得进一步研究。本实验设计合成的2-乙基-6-甲基-3-烷基-3H-喹唑啉-4-酮类化合物是一类全新结构的喹唑啉酮类化合物,为进一步研究喹唑啉酮类化合物的结构优化和构效关系奠定了基础,利于喹唑啉酮类化合物合成工艺的放大研究。
喹唑啉酮类化合物具有较为广泛的生物活性,利用喹唑啉酮类先导化合物进行结构修饰,不断优化该类化合物的合成工艺,继续深入研究其生物活性,将是此类化合物研究的重要方向。
[1] 刘 刚,李晓燕.喹唑啉酮类化合物合成新方法研究进展[J].药学进展,2007,31(12):542-550.
[2] 佟茂国.喹唑啉类化合物的合成及其抗肿瘤活性研究进展[J].广州化工,2012,40(3),17-19.
[3] Chand rika P M,Yakaiah T,Gayatri G,et al.Click chem istry:studies on the synthesis of novel fluorous tagged triazol-4-yl substituted quinazoline derivatives and their biological evaluation-theoretical and experimental validation[J].Eur J M ed Chem,2010,45:78-84.
[4] El-Gazzar ABA,Youssef MM,Youssef AM S,et al.Design and synthesis of azolopyrim idoquinolines,pyrim idoquinazolines as anti-oxidant,anti-inflammatory and analgesic activities[J].Eur JMed Chem,2009,44:609-624.
[5] 李文举,欧阳贵平,张广龙.4-取代氨基喹唑啉类化合物的研究进展[J].精细化工中间体,2009,39(3),15-20.
[6] Lee JY,Park YK,Seo SH,et al.1,4-dioxane-fused 4-anilinoquinazoline as inhibitors of epidermal grow th factor receptor kinase[J].A rch Pharm Res,2001,334(11):357-360.
[7] Lee JY,Lee YS,Park HK,et al.4-(Phenylam ino)[1,4]dioxano[2,3-g]quinazoline derivatives and process for preparing the same:USA,2003045537[P].2003-03-06.
[8] 宁微微,刘雪飞,张晓梦,等.新型喹唑啉酮类先导物的设计、合成及抑制人顶体酶活性研究[J].药学实践杂志,2010,28(4),296-298,312.
[9] Ioannis K,Abdelhakim E,Elisabeth S,et al.Rapid synthesis of 2,3-disubstituted-quinazolin-4-ones enhanced by m icrow ave-assisted decom position of formam ide[J].Tetrahedron Lett,2007,48:6609.
[10] 宋桂红,张 珏,张晓梦,等.南德士抑制人精子顶体酶活性的实验研究[J].中华男科学杂志,2009,15(8):700.
[11] 付丙月,唐 辉,郑灿辉,等.四氢萘类化合物合成工艺研究[J].药学实践杂志,2012,30(1),35-37.
[12] NCCLS.Reference method for broth dilution antifungal susceptibility testing of yeasts;approved standard[S].2nd ed.Villanova:[s.n.],2000.
[13] 曹永兵,姜远英,王 宁,等.微量液基稀释法测定化合物特苄康唑的体外抗真菌活性[J].中国抗生素杂志,2000,25,183.
Synthesis of substituted novel quinazolinone compounds
ZHAO Lei1,2,ZHAO Xuemei1,TANG Hui1,WU Juan3(1.Shandong Provincial Hospitalaffiliated to Shandong University,Jinan 250021,China;2.Qilu Hospital of Shandong University,Jinan 250012,China;3.Chengdu M ilitary General Hospital,Chengdu 610083,China)
ObjectiveTo optimize the synthesis route of substituted quinazolinone and investigate the influence of temperature and time in the synthesis of key intermediates.MethodsA parallel testwas carried out to compare temperature,time and the ester bond ammonolysis of condensing agent,including the totalanalysis of experimental result.And orthogonalexperimental design was used and the influence of temperature and time on the yield were investigated.Results and ConclusionThe structure of the compound was confirmed by1H NMR,MSand13C NMR.When the reaction temperaturewas 30℃,the reaction timewas 24h with the condensing agent(DCC)and the reactant ratio of 1.5∶1,the yield of ester bond ammonolysiswas higher.The optimal preparation procedure of quinazolinone compounds wasmore available for industrial production.In vitro antifungal activity test results showed that the lead compoundsmeasured on five clinical pathogenic fungihave the potential antifungal activity,and are worth for further study.
quinazolinone compound;synthesis;orthogonal design;analysis of variance
O621.3
A
1006-0111(2015)06-0529-04
10.3969/j.issn.1006-0111.2015.06.013
2014-05-19
2014-11-06[本文编辑]陈静
山东省自然科学基金(No.ZR2012HQ026);成都军区总医院2015年院管课题
赵 蕾,硕士研究生.研究方向:中药新制剂与临床药学.Tel:(0531)68776465;E-mail:165703687@qq.com
唐 辉,博士.研究方向:新药研究与药物质量安全研究.Tel:(0531)68776449;E-mail:tanghui1110@163.com
