HPLC法同时测定氯霉素氢化可的松滴耳液中两组分含量
2015-05-21罗自力杜建红何德云钟元高申冬妮
祝 辉,罗自力,杜建红,何德云,钟元高,申冬妮,李 佳
氯霉素氢化可的松滴耳液是常用医院制剂,收载于《中国医院制剂规范》西药制剂(第二版)[1]。其主要成分为氯霉素和氢化可的松,具有抗炎、抗过敏作用,临床用于中耳炎的治疗。现行检验标准采用紫外分光光度法测定制剂中氯霉素含量,却未对氢化可的松含量作具体规定。文献报道采用HPLC法同时测定氯霉素氢化可的松滴耳液中两组分含量[2-4],所选用的流动相为不同比例甲醇-水,氯霉素和氢化可的松在5 min内先后出峰,特别是氯霉素出峰时间约3 min,易受溶剂峰干扰。本研究参考《中国药典》2010版(二部)中氯霉素和氢化可的松原料药及其制剂含量测定方法[5-6],以离子对试剂——庚烷磺酸钠为流动相调节剂,建立了同时检测氯霉素和氢化可的松含量的HPLC方法,并进行了方法学验证,为制剂标准提高提供参考。
1 仪器与试药
1.1 仪器 高效液相色谱仪(Agilent1100,美国),优普系列超纯水器(UPT-Ⅰ,成都超纯科技有限公司),超声波清洗器(KQ5200E,昆山市超声仪器有限公司),电子分析天平(BT25S,赛多利斯,德国)。
1.2 试药 氯霉素对照品(130555-200602,中国药品生物制品检定所,纯度99.4%),氢化可的松对照品(100152-200602,中国药品生物制品检定所,纯度:100%),庚烷磺酸钠(20130316,成都市科龙化工试剂厂),二甲基甲酰胺(20110309,重庆中渝化工试剂厂),冰醋酸(20130606,成都市科龙化工试剂厂),乙腈(A998-4,Fisher公司),超纯水(自制),氯霉素氢化可的松滴耳液(批号:130820A、130911A、131025A,某医院自制)。
2 方法与结果
2.1 溶液制备
2.1.1 混合对照品溶液制备 精密称取氯霉素对照品 10.20 mg、氢化可的松对照品 10.11 mg,分别置10、50 ml量瓶中,加水溶解,稀释并定容至刻度,摇匀,作为对照品储备液A、B;再分别精密量取储备液A 1 ml,储备液 B 1 ml置10 ml量瓶中,加水稀释并定容至刻度,摇匀,即得。
2.1.2 供试品溶液制备 精密量取氯霉素氢化可的松滴耳液1 ml置50 ml量瓶中,加水稀释,定容至刻度,摇匀,即得。
2.1.3 阴性对照溶液制备 按照处方工艺制备阴性对照,参照“2.1.2”项下制备成溶液。
2.2 色谱条件与系统适应性实验 色谱柱:WondaSil色谱柱(C18-WR;4.6 mm×250 mm;5 μm);流动相:乙腈-0.1%庚烷磺酸钠溶液(取0.1%的庚烷磺酸钠溶液500 ml与二甲基甲酰胺5 ml、冰醋酸0.5 ml混匀,超声脱气 5 min,即得)(26∶74),检测波长 :0~16 min 为 278 nm,16~20 min 为 245 nm,20~25 min 为 278 nm;柱温:30 ℃;流速:1.0 ml/min;进样量:20 μl。系统适应性实验结果表明:阴性对照在氯霉素和氢化可的松出峰处无干扰(图1)。
2.3 方法学考察
2.3.1 线性关系考察 精密吸取“2.1.1”项下对照品溶液 10、15、20、25、30、40 μl,分别注入 HPLC 仪,记录出峰时间与峰面积,以峰面积(Y)为纵坐标,进样量(X,μg/ml)为横坐标进行线性回归,得氯霉素线性回归方程式为 Y=27.021X+9.71,r=1 (n=6);氢化可的松线性回归方程式为 Y=40.867X-7.53,r=0.9999(n=6)。 结果表明:氯霉素在 4.08~204 μg/ml,氢化可的松在 10.11~40.44 μg/ml范围内,与峰面积均呈良好的线性关系。
2.3.2 重复性试验 取批号为 130820A样品 6份,按“2.1.2”供试品溶液制备方法得供试品溶液,照“2.2”项下色谱条件下进行含量测定,记录峰面积。结果:氯霉素、氢化可的松峰面积RSD分别为0.5%、0.6%(n=6),表明该方法重复性试验好。

图1 高效液相色谱
2.3.3 精密度试验 取“2.1.1”项下对照品溶液,按照“2.2”项下色谱条件依次连续进样5次,每次20 μl,记录峰面积,结果:氯霉素、氢化可的松峰面积的RSD分别为0.5%、0.7%(n=5),表明该方法精密度试验好。
2.3.4 稳定性试验 取批号为130820A样品稀释溶液, 室温下存放,1 d 内依次在第 2、5、8、12、16、20、24 h,按“2.2”项下色谱条件进样,记录峰面积,结果:氯霉素、氢化可的松峰面积的RSD分别为0.9%、1.0%(n=7),表明该样品在 24 h内稳定。
2.3.5 加样回收试验 精密量取已知含量的样品9份(批号:130820A,含氯霉素和氢化可的松分别为2.505 mg/ml、0.512 mg/ml), 每 份 1 ml, 按 80% 、100%、120%比例,分别加入氯霉素和氢化可的松对照品适量,照“2.1.2”供试品溶液制备方法稀释至刻度,摇匀,按“2.2”项下色谱条件进行含量测定,计算回收率。结果见表1。
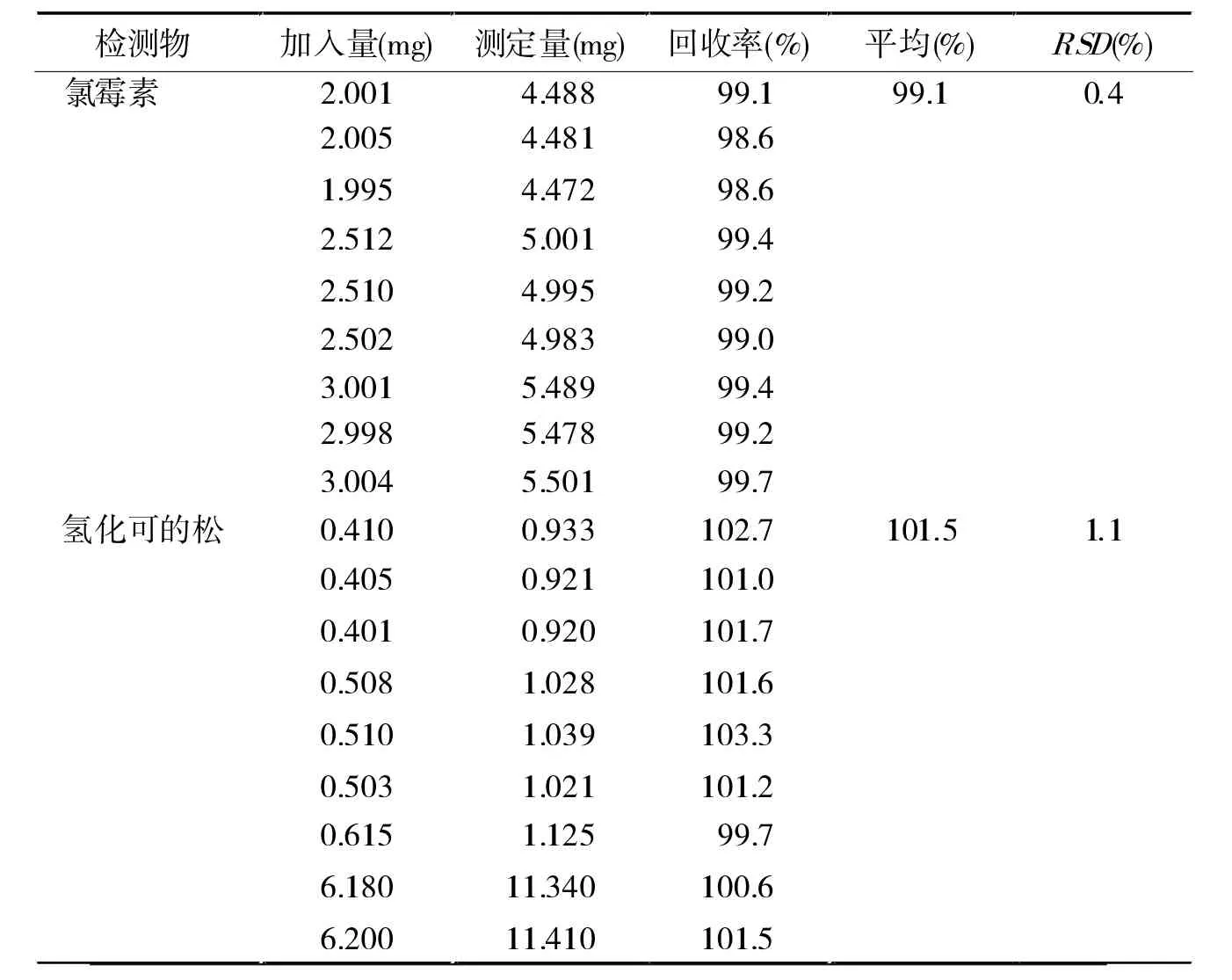
表1 加样回收率试验结果(n=9)
2.4 样品含量测定 将130820A、130911A、1310 25A 3个批次样品按照“2.1.2”方法处理,每个批次平行取2份,制备供试品溶液;再精密称取对照品2份,照“2.1.1”方法处理制备对照品溶液,每份供试品溶液及对照品溶液按照“2.2”项下色谱条件方法依次进样2次,记录峰面积,按外标法计算样品百分含量,结果见表2。
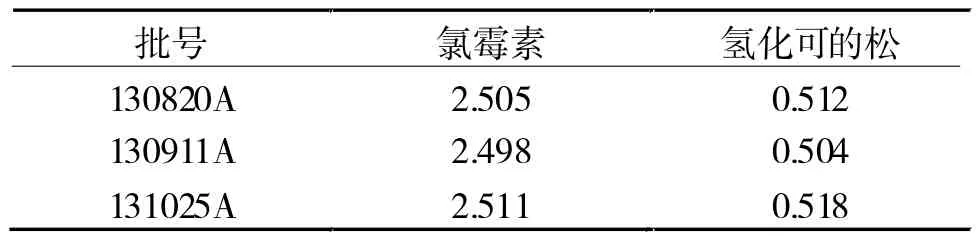
表2 氯霉素氢化可的松滴耳液的含量测定结果(mg/mI)
3 讨论
氯霉素氢化可的松滴耳液来源于医院制剂,主要成分为氯霉素和氢化可的松,现行质量标准采用紫外分光光度计法测定氯霉素含量,并未对氢化可的松含量做具体规定,不能对该药品进行全面质量监控,质量标准有一定局限性。已有文献报道采用HPLC方法同时检测氯霉素和氢化可的松含量,但文献方法中两组分出峰时间短,特别是氯霉素出峰时间过快(约3 min),存在溶剂峰干扰。本研究选择庚烷磺酸钠作为离子对调节剂,有效提高了两组分保留时间,出峰时间优化为15 min左右,经方法学验证,新建HPLC方法适合用于同时检测氯霉素和氢化可的松含量。
检测波长的选择:以流动相为空白,在200~400 nm波长范围内,扫描氯霉素和氢化可的松紫外吸收,结果显示氯霉素、氢化可的松分别在278 nm、245 nm有最大吸收峰。因此,本研究设定初始检测波长278 nm,待氯霉素出峰之后,变换检测波长为245 nm,有效提高了对氯霉素和氢化可的松的检测灵敏度。
[1]中华人民共和国卫生部药政局.中国医院制剂规范(西药制剂第二版)[S].北京:中国医药科技出版社,1995:158-159.
[2]李小燕.HPLC法测定氯霉素氢化可的松滴耳液中两组分的含量[J].色谱,1998,16(1):71-73.
[3]陈豪,刘芳群,曾建国,等.HPLC法测定氯霉素氢化可的松滴耳液中两组分的含量[J].中国药师,2010,4(13):585-586.
[4]金克林.高效液相色谱法测定氯霉素氢化可的松滴耳液中氯霉素和氢化可的松的含量[J].中国现代应用药学杂志,2011,6(20):507-508.
[5]国家药典委员会.中华人民共和国药典(二部)[S].北京:中国医药科技出版社,2010:1029-1032.
[6]国家药典委员会.中华人民共和国药典(二部)[S].北京:中国医药科技出版社,2010:550-552.
