UPLC法测定炮制前后王不留行中Vaccarin含量
2015-04-26阴钺玲孙兆林
阴钺玲,薛 睿,孙兆林*
(1.哈尔滨商业大学 生命科学与环境科学研究中心,黑龙江 哈尔滨150076; 2.中国科学院上海药物研究所,上海 201203)
UPLC法测定炮制前后王不留行中Vaccarin含量
阴钺玲1,2,薛 睿2,孙兆林2*
(1.哈尔滨商业大学 生命科学与环境科学研究中心,黑龙江 哈尔滨150076; 2.中国科学院上海药物研究所,上海 201203)
目的:考察炮制前后王不留行中Vaccarin的含量变化。方法:采用 UPLC 法测定王不留行中Vaccarin的含量,色谱柱为BEH C18( 100mm ×2.1mm,1.7μm),以0.3%磷酸水溶液(A)-乙腈(B)为流动相,流速为0.2mL·min-1,检测波长为280nm,梯度洗脱(0~4min, 10%→20%B;4~10min,20%→45% B),柱温为35℃。结果:Vaccarin在生品王不留行中含量较高,而炮制后其含量大幅降低。结论:炮制前后王不留行中Vaccarin的含量可发生明显变化,临床用药时需谨慎选择。
王不留行;Vaccarin;UPLC;炮制;含量变化
王不留行为石竹科麦蓝菜Vaccariasegetalis(Neck.)Garcke的干燥成熟种子,又名奶米、王不留、大麦牛、麦蓝子。其在国内分布广泛,以华北、东北为主,主治下乳消肿、经闭、痛经、乳痈肿痛等[1]。王不留行的主要成分为环肽、三萜皂苷、黄酮类化合物等[2-4]。《中国药典》(2010年版)中收载的王不留行包括生品和炒制品两种,目前生品应用较为广泛,但是对生品和炮制品的区别尚未有研究报道。Vaccarin是《中国药典》中规定的王不留行药材质量评价的指标成分。因此,本文拟采用超高效液相色谱(UPLC)法,考察炮制前后王不留行中Vaccarin的含量变化,拟从化学成分含量变化的角度对生品和炒制品的区别进行研究。
1 仪器与试剂
Waters AcquityTM Ultra Performance Liquid Chromatography(UPLC)色谱仪,包括四元泵、脱气机、二极管阵列紫外检测器、自动进样器、柱温箱、Empower 2色谱工作站(美国Waters公司);分析天平(瑞士梅特勒-托利多集团公司);SK3300型超声波清洗器(上海科导有限公司)。
Vaccarin对照品(批号MUST-15012111,成都曼思特生物科技有限公司)。生、炒王不留行药材均为市售(同批为生、炒药材各1份,北京同仁堂药店,批号:20150120、20150209、20150311)。
乙腈、甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 对照品溶液配制
精确称定对照品Vaccarin 5.1mg, 加70%甲醇溶解并定容至10mL容量瓶中,摇匀,制得对照品储备液。
2.2 供试品溶液配制
精密称取生、炒王不留行药材各700mg,置于具塞锥形瓶中,精密加入70%甲醇25mL,称定重量,超声(180W,35kHz)提取45min,放冷,再称定重量,补足减失的重量,摇匀,用0.22μm微孔滤膜滤过,取续滤液,即得。
2.3 色谱条件
BEH C18色谱柱( 100mm ×2.1mm,1.7μm),流动相:0.3%磷酸水溶液(A)-乙腈(B),梯度洗脱 (0~4min,10%→20% B; 4~10min,20%→45%B) ,流速:0.2mL·min-1,检测波长:280nm,柱温:35℃,进样: 5.0μL。
2.4 系统适用性试验
在“2.3”项色谱条件下,分别取空白样品溶液、对照品溶液和供试品溶液各5.0μL进样检测,结果表明Vaccarin与其相邻色谱峰分离度均大于1.5,理论塔板数不低于3 000,拖尾因子均在0.95~1.05之间,峰形良好。见图1。
2.5 线性关系考察
精密吸取对照品储备液1.0mL,置于10mL容量瓶中,加70%甲醇定容至刻度,摇匀。按照“2.3”项下色谱条件进行测定,进样体积分别为 1.0、3.0、5.0、8.0、10.0μL,以Vaccarin对照品的进样量(X)为横坐标,对应色谱峰峰面积(Y)为纵坐标,绘制标准曲线计算得回归方程。见表1。
2.6 精密度试验
精密吸取“2.5”项下对照品溶液和生、炒供试品溶液,按照“2.3”项下色谱条件连续进样测定6次,每次进样5.0μL,结果表明Vaccarin峰面积RSD (n=6)分别为0.43%、0.69%、0.58%,表明仪器精密度良好。
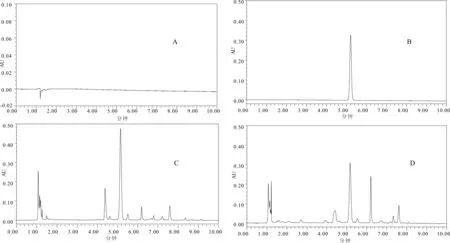
图1 280nm处空白样品溶液(A)、对照品溶液(B)、生王不留行样品溶液(C)、炒王不留行样品溶液(D)UPLC色谱

组分回归方程r线性范围(μg·μL⁃1)LODLOQVaccarinY=8000000X-1789840.99930.051~0.510.0150.051
2.7 稳定性试验
分别取“2.2”项下对照品溶液和生、炒供试品溶液,于配制后0、2、4、8、12、24h分别进样5.0μL,按照“2.3”项下色谱条件进行测定,记录色谱峰峰面积,计算得RSD (n=6)分别为0.62%、0.77%、0.74%,表明供试品溶液在24h内稳定。
2.8 重复性试验
取20150120批次的生、炒王不留行药材,平行样品各6份,按照“2.2”项下方法制备供试品溶液,分别进样,按照“2.3”项下色谱条件测定。结果表明样品中Vaccarin的含量平均值(n=6)为24.43%、31.79%,其RSD为1.4%、1.1%,表明该方法重复性良好。
2.9 回收率试验
准确称取已知含量的上述生、炒王不留行药材各6份,按“2.2”项下方法制得供试品溶液,分别精密加入Vaccarin对照溶液各5μL,按“2.3”项下色谱条件进样测定,计算得生、炒王不留行的平均回收率分别为98.4%、99.1%,RSD为0.77%、0.45%。
2.10 样品测定
取不同批次的3批样品,按照“2.2”项下方法分别制备3份供试品平行溶液,进样5.0μL,记录峰面积,计算其含量平均值,结果见表2(A:生王不留行,B:炒王不留行)。
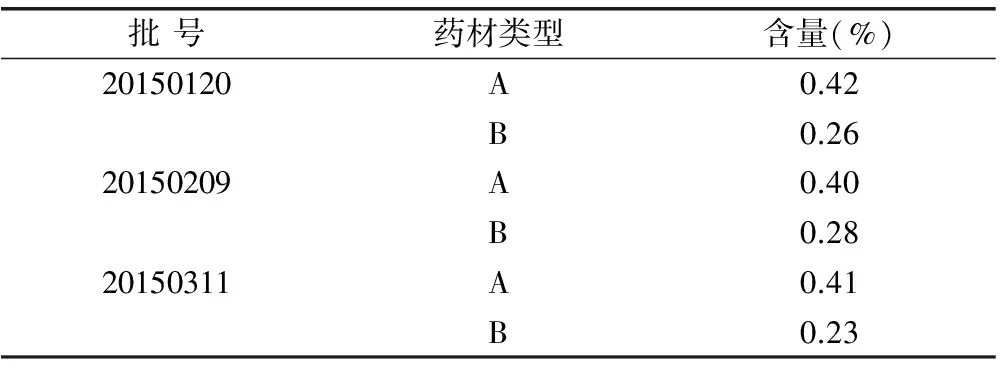
表2 样品含量测定结果
3 讨论
UPLC具有检测快捷、操作方便、准确度高、重复性好的特点,现已广泛应用于植物、矿物等天然中药化学成分的分析,可为中国药典中药品快速检验技术的改进提供科学参考[5]。
本文建立了UPLC测定王不留行中Vaccarin含量的方法,经系统方法学验证表明该方法稳定可行。利用所建立的方法对炮制前后的王不留行中Vaccarin的含量变化进行考察,结果表明,炮制前后的3批药材中Vaccarin的平均含量分别为0.41%和0.26%。《中国药典》中规定王不留行生品和炒制品中Vaccarin含量分别应不低于0.40%和0.15%,因此本文中所测王不留行均符合药典标准。
根据中药炮制理论,种子类药材在炒制后致使皮破裂而利于成分煎出,因此药材炮制后的有效成分含量应高于未炮制药材,然而本文却有截然相反的发现。推测其原因可能是Vaccarin是黄酮苷类化合物,其在高温条件下稳定性较差。据文献报道,侧柏叶黄酮类化合物在50℃以上的环境中稳定性明显下降[6];槲树叶黄酮类化合物在80℃以上的环境中含量可随温度升高而逐渐下降[7]。因此,在本研究中,王不留行经炒制后Vaccarin含量明显下降,可能是由于药材经高温炒制导致黄酮类成分破坏流失,但具体机制还需进一步研究。
[1] 国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社,2010:49-50.
[2] 桑圣民,劳爱娜,王洪诚,等.中药王不留行化学成分的研究(Ⅱ)[J].中草药,2000,31(3):11-13.
[3] 桑圣民,毛士龙,劳爱娜,等.中药王不留行化学成分的研究Ⅲ[J].天然产物研究与开发,2000,12(3):12-15.
[4] 桑圣民,夏增华,毛士龙,等.中药王不留行中黄酮甙类成分的研究[J].中国中药杂志,2000,25(4):29-30.
[5] 刘明理,马双成.药品快速检验技术研究及应用管理中需要加强的工作[J].中国药事,2012,26(8):857-858, 870.
[6] 赵莹,张建平,赵永光,等.侧柏叶黄酮类化合物的稳定性[J].河北科技师范学院学报,2008,22(3):30-33.
[7] 南海娟,郭延成,颜振敏,等.槲树叶黄酮类化合物的提取及稳定性[J].食品与发酵工业,2013,39(4):234-237.
(责任编辑:尹晨茹)
2015-06-09
国家重大新药创制科技重大专项(2012ZX09301001)
阴钺玲(1989-),女,哈尔滨商业大学硕士研究生,研究方向为中药药物分析。
孙兆林(1980-),中国科学院上海药物研究所中级实验师,研究方向为中药质量控制。
R284.2
A
1673-2197(2015)20-0020-02
10.11954/ytctyy.201520009
