基于相容性选择NEPE推进剂固化剂的分子模拟
2015-04-22侯世海武文明李红霞
王 广,侯世海,武文明,李红霞
(第二炮兵工程大学601室,西安 710025)
基于相容性选择NEPE推进剂固化剂的分子模拟
王 广,侯世海,武文明,李红霞
(第二炮兵工程大学601室,西安 710025)
构建了环氧乙烷-四氢呋喃共聚醚(PET)及常用固化剂多官能度异氰酸酯(N-100)、甲苯二异氰酸(TDI)和异佛尔酮二异氰酸(IPDI)的分子模型及无定形结构,采用分子动力学方法计算了这4种组分的溶度参数,并对组分纯物质间及混合体系组分间的径向分布函数进行了分析,采用共混方法计算了不同固化剂分别与PET组成的共混体系的共混能,得到了不同共混体系的共混结合能分布图。分析结果得到一致结论,PET与固化剂相容性优劣次序为PET/N-100>PET/IPDI>PET/TDI。结论与目前工程应用中普遍采取N-100作为NEPE推进剂的固化剂这一实际相吻合,验证了采用分子模拟方法从相容性能选择固化剂的可行性,该方法可预测不同组分的相容性,为固体推进剂的配方设计提供参考。
NEPE推进剂;固化剂;相容性;分子模拟
0 引言
目前,在NEPE推进剂体系中广泛使用的是聚氨酯粘合剂,虽然只占总质量的6%~8%,但却起着重要的作用。这类粘合剂主要成分有环氧乙烷-四氢呋喃共聚醚(PET),由于这种共聚醚主链结构的不规整性以及醚键的存在,使其具有不易结晶、力学和工艺性能好的特点,是NEPE推进剂的一种较为理想的聚醚多元醇。工程应用中,向粘合剂中加入一定量的固化剂,使低分子聚合物或单体经一定的化学反应生成高分子化合物,或使线型高分子化合物交联成体型高分子化合物[1]。聚氨酯粘合剂中采用的固化剂主要为多官能度异氰酸酯(N-100)、甲苯二异氰酸(TDI)、异佛尔酮二异氰酸(IPDI)等,固化剂的选择不仅要注重交联固化的效果,还要与其他组分具有很好的相容性,这也是固体推进剂成型过程得以实现的前提[2]。相容性直接影响着推进剂的力学性能,精确地表征相容性是研究推进剂共混体系的基础[3],实验方法过程复杂,且耗时耗力。分子模拟技术克服了实验研究的缺点,成为研究固体推进剂等含能材料微观性质的一种非常有效的手段。
焦东明[4]采用分子动力学(MD)方法计算了丁羟推进剂粘合剂、增塑剂的溶度参数,来考察组分间的相容性,模拟数值与实验基本一致。兰艳花等[3]应用MD方法模拟计算了HTPB固体推进剂粘合剂与增塑剂组分DOS、DOA、DBP和DOP所组成共混物的密度、结合能和径向分布函数。以及纯物质的溶度参数,并根据模拟计算结果,评价了HTPB与不同增塑剂相容性的优劣,次序为HTPB/DOS>HTPB/DOA>HTPB/DOP>HTPB/DBP;黄锐、姚维尚等[5-8]对纤维素基、聚酯类等不同粘合剂进行了模拟计算,为制得力学性能较好的高能粘合剂提供了依据;杨月诚[9]对HTPB粘合剂及其常用的增塑剂、固化剂组分的溶度参数进行了模拟计算,发现相容性对固化效果有一定影响。但以上研究所建模型采用的原子数量较少,计算结果精度较低,且分析方法较单一,不能直观展示组分相容性。
本文采用MD方法对PET和不同固化剂的纯物质及其组成的共混体系进行了模拟计算,针对以上不足,建模时适当增加模型原子数,对溶度参数模拟进行了验证,提高了计算结果的准确性,在分析径向分布函数的基础上,增加了Blends分析方法,不仅计算出混合能,而且通过结合能分布图更加直观地展示组分相容性。运用MD方法很容易得到组分溶度参数,而径向分布函数分析法和Blends方法的结果更加直观,本文使用这3种分析方法,可从不同的角度去分析组分相容性,其模拟结果能够为NEPE推进剂及其他含能材料的配方设计提供参考依据。
1 基本理论与方法
1.1 理论依据
从热力学观点来看,2种物质混合时,有以下热力学关系[10]:
ΔGM=ΔHM-TΔSM
(1)
ΔHM=ΔEM+pΔVM
(2)
式中ΔGM、ΔHM、ΔEM和ΔSM分别为Gibbs混合自由能、混合焓、混合能和混合熵;T、p和ΔVM分别为混合时的温度、压力和体积变化。
两组分能否相容并得到均相共混物,取决于ΔGM,这里讨论的相容性是热力学相容性,即分子水平上的均相体系。只有满足ΔGM<0时,两组分互容,且溶解过程能够自发进行;溶解中熵总是增加的,即ΔSM>0,对于推进剂组分内弱极性或非极性高分子体系的混合溶解过程一般是吸热过程,即ΔHM>0;为了满足ΔGM<0,ΔHM值越小或越趋近于零较好。
根据Hildebrand的半经验公式[11]:
ΔHM=VMφ1φ2(δ1-δ2)2
(3)
式中VM为总体积;φ1和φ2是2种组分的体积分数;δ1和δ2是2种组分的溶度参数。
从式(3)可看出,δ1和δ2的差越小,则ΔHM值越小,越有利于溶解,这就是溶度参数相近原则。实验表明,对非晶态高分子材料来说,若分子间没有强极性基团或氢键基团,2种组分只要满足|δ1-δ2|<1.7~2.0(J/cm3)1/2[12],则2种组分相容。
1.2 模拟方法
分子动力学(MD)方法通过计算分子的汽化热预测材料的溶度特性,首先使用Materials Visualizer构建分子的空间构型,采用Discover模块中的Smart Minimizer进行几何优化,而后构建其无定形体,并进行优化,优化方法采用智能最小优化方法,在正则系综(NVT)条件下,进行250 ps的分子动力学模拟,前200 ps用于平衡,后50 ps用于计算。动力学运算后,采取Analysis来获取溶度参数,分析径向分布函数,判断相容性。其中,力场采用COMPASS力场,能量最优化采用最陡下降法(Steepest Descent)和共轭梯度法(Conjugate Gradient)[13],温度控制用Anderson[14]方法,非键截断(cutoff)取0.95 nm,样条宽度(spline width)取0.1 nm,缓冲宽度(buffer width)取0.05 nm,时间步长1 fs,静电和范德华力计算采取Eward[15]和Atom-based[16]长程加和方法。
共混(Blends)方法是通过Monte Carlo模拟后加权分子空间取向排布计算结合能来判断组分相容性,将优化后的不同组分的分子模型设置为screen和base作为共混模块输入条件,在COMPASS力场条件下,提交到mixing任务进行共混模拟,计算各个组分的结合能,通过分析共混结合能分布曲线,判断组分相容性。
1.3 方法验证
使用MD方法模拟计算常见聚合物的溶度参数,以聚乙烯(PE)、聚丙烯(PP)和端羟基聚丁二烯(HTPB)为例,聚合物的物理性质(298 K)和计算出的聚合物溶度参数见表1。

表1 MD 方法模拟计算结果验证Table1 Confirmation of MD simulation results
将MD模拟计算的结果δMD与实验数据δexp及焦东明[9]采用的Synthia方法模拟计算得到的结果δSynthia进行对比。可看出,MD方法得到的数值较好吻合实验值和采用Synthia方法得到的模拟值,且误差在允许范围内。因此,可用来模拟计算组分的溶度参数,进而研究组分相容性。
2 模拟计算与结果分析
2.1 模型构建
运用MS软件中的Visualizer模块,根据PET、N-100、TDI、IPDI的分子简式建立相应的分子模型,如图1所示。利用Amorphous cell模块,构建上述4种组分的纯物质以及PET与固化剂混合体系的无定形分子模型。模型中原子总数约为1 000个。

图1 PET、N-100、TDI、IPDI分子模型Fig.1 Molecule model of PET,N-100,TDI and IPDI
2.2 溶度参数分析方法
对4种组分纯物质的无定形模型进行优化后,进行分子动力学运算并进行分析,组分的物理性质及模拟计算,结果见表2。从表2可看出,对PET、N-100、TDI、IPDI溶度的模拟,能够定量地得到溶度参数。
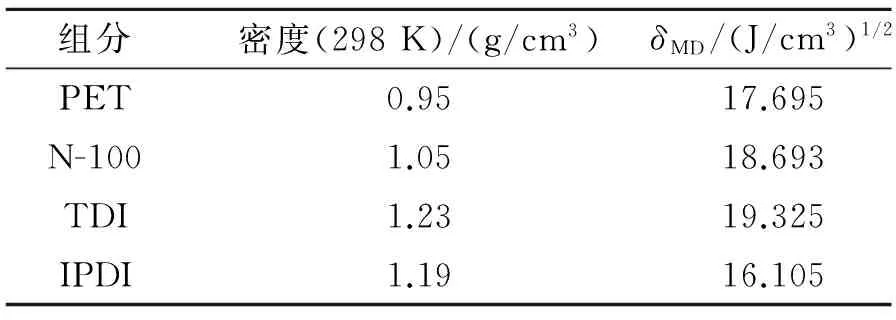
表2 MD方法模拟计算结果Table2 MD simulation results
4种组分的溶度参数值均较接近,经过计算得到溶度参数差值见表3。从表3可看出,PET与3种固化剂的溶度参数差值的绝对值均小于1.7(J/cm3)1/2,根据溶度参数相近原则,说明PET与这3种固化剂能很好的相容,且PET与N-100的相容性好于PET与IPDI的相容性,PET与IPDI的相容性好于PET与TDI的相容性。
2.3 径向分布函数分析方法
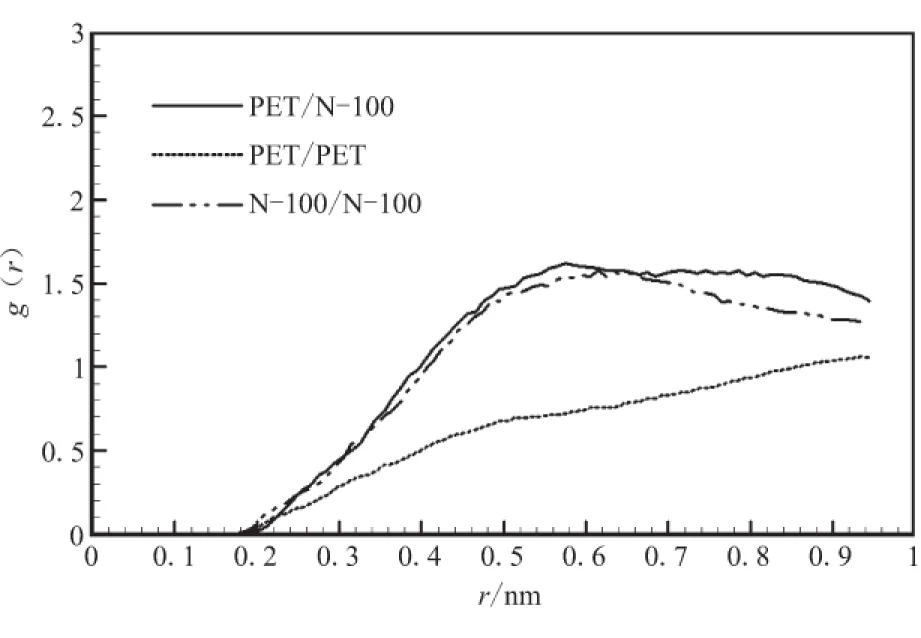
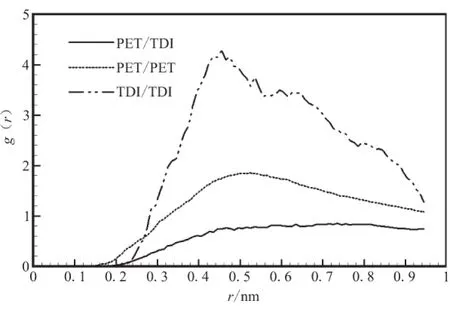
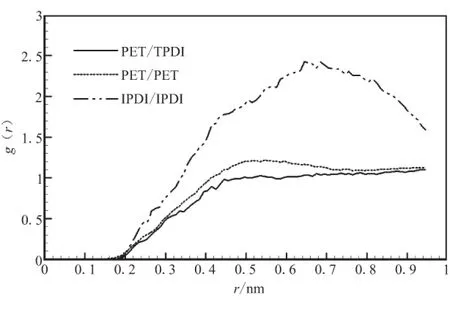
对PET与3种固化剂组成的共混物进行优化,并进行MD运算后,分析其径向分布函数g(r)。g(r)是反映材料微观结构的特征物理量,它表示在一个分子周围距离为r的地方出现另一个分子的概率密度相对于随机分布概率密度的比值。分子间径向分布函数可揭示非键原子间相互作用的方式和本质,氢键作用范围为0.26~0.31 nm,范德华力(van der Waals, vdw)作用范围为0.31~0.50 nm[17],从图2可看出,4种纯物质分子间主要作用方式为vdw作用,PET与3种固化剂的分子间径向分布函数曲线在vdw作用范围内距离越近,说明PET与之相容性好,在vdw作用范围内,距离PET最近的是N-100,其次是IPDI,最远的是TDI,说明固化剂与PET相容性优劣顺序为N-100>IPDI>TDI。
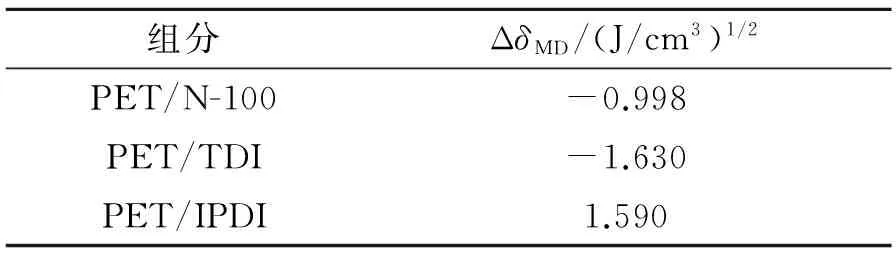
表3 组分溶度参数差值Table3 ΔδMD of different components

图2 纯物质分子间径向分布函数Fig.2 Intermolecular pair correlation functions for purePET,N-100,TDI and IPDI
对于混合体系而言,分子间径向分布函数也可用来判断混合物的相容性。混合物的分子间径向分布函数g(r)越是高于单一物质的径向分布函数,相容性越好;反之,则会发生相分离[3]。
2.4 Blends分析方法
在Blends模块中,将PET分子模型设置为base,将N-100、TDI和IPDI设置为screen,分别组成共混体系,提交Mixing任务进行计算,得到混合能如表4所示,共混结合能分布曲线如图4所示。
(a)PET/N-100
(b)PET/TDI
(c)PET/IPDI
从图3可看出,混合体系分子间主要作用方式也是vdw作用,在PET / N-100混合体系中,PET-(N-100)的分子间g(r)值高于(N-100)-(N-100)和PET-PET的分子间g(r)值,而在PET/TDI混合体系和PET/IPDI混合体系中同组分的分子间g(r)值均高于两组分分子间g(r)值,但在PET/TDI混合体系中,g(r)差值较大,故可判断相容性结论为PET/N-100>PET/IPDI>PET/TDI。

表4 Blends方法模拟计算混合能Table4 Mixing energy calculated by Blends method
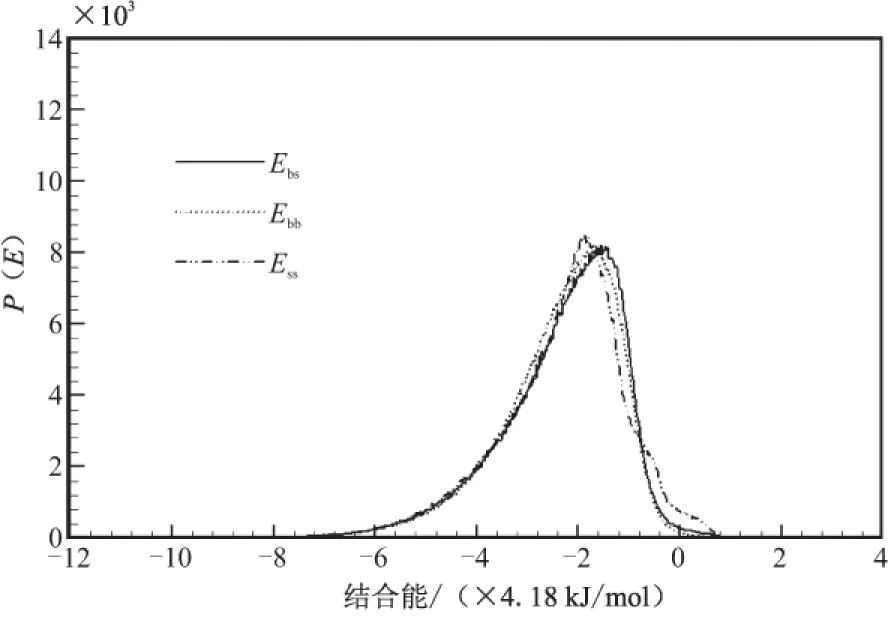
(a)PET/N-100
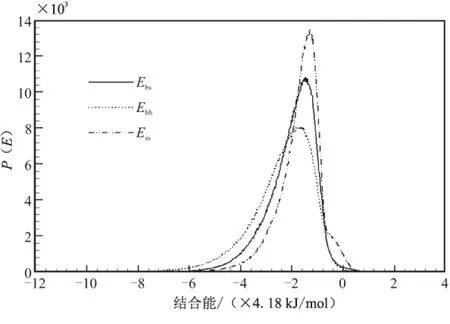
(b)PET/TDI
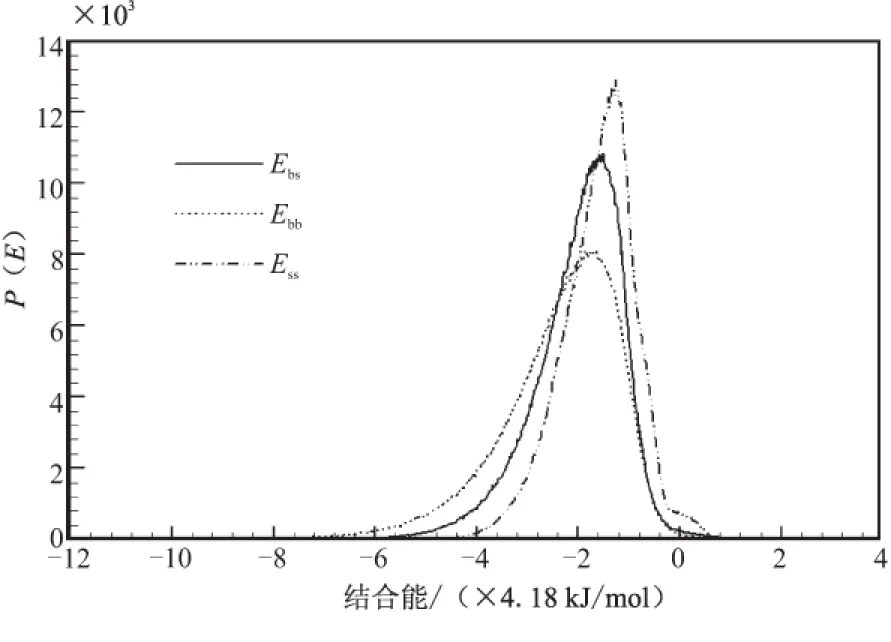
(c)PET/IPDI
从计算结果看出,3种不同共混体系的混合能关系EPET/N-100 混合能与组分间吸附能满足以下关系式[18]: (4) 式中Eij为分子间的吸附能(Binding energy);Z为配位数(Coordination numbers);MS软件Blends模块通过设定组分为base和screen角色,分别计算base-base、screen-screen和base-screen组分的吸附能,即Ebb、Ess和Ebs(Ebs=Esb)。 从图4可看出,PET/N-100共混时,共混结合能Ebb、Ess和Ebs这3个值分布较接近,其次是PET/IPDI共混体系、PET/TDI共混体系,共混结合能分布越接近,体系的混合能就越小。所以,可得出PET与3种固化剂的相容性优劣顺序为PET/N-100>PET/IPDI>PET/TDI。 (1)PET与固化剂N-100、TDI及IPDI的相容性优劣顺序为PET/N-100>PET/IPDI>PET/TDI。从相容性角度考虑,NEPE推进剂粘合剂中,选用N-100作为固化剂,效果较好。 (2)溶度参数法简单易实现,径向分布函数分析与Blends方法显示结果直观明了,这3种方法能够从不同角度去分析组分相容性,综合运用这3种方法,可准确直观地判断组分间相容性。 (3)本文模拟方法可作为预测NEPE推进剂其他组分相容性的有利工具,也可为固体推进剂和炸药的配方设计提供参考依据。 [1] 徐婉.NEPE推进剂固化体系研究[D].长沙:湖南大学,2009. [2] 齐晓飞,张晓宏,宗振伟,等.分子动力学方法在火炸药研究中的应用进展[J].化学推进剂与高分子材料,2012,10(3):37-42. [3] 兰艳花,刘亚青,付一政.HTPB与增塑剂相容性评价的分子动力学模拟[J].含能材料,2010,18(1):42-46. [4] 焦东明,杨月诚,强洪夫,等.HTPB固体推进剂增塑剂选取分子模拟研究[J].化学研究与应用,2009,21(6):805-809. [5] 黄锐.叠氮纤维素的分子模拟研究[D].北京:北京理工大学,2008. [6] 黄锐,姚维尚,谭惠民.叠氮纤维素结构和溶度参数的分子模拟[J].含能材料,2008,16(4):446-449. [7] 黄锐,姚维尚,谭惠民.纤维素基含能黏合剂的分子模拟[J].火炸药学报,2008,31(1):64-67. [8] 姚维尚,李倩,谭惠民.NEPE推进剂粘合剂性能的分子模拟研究[J].含能材料,2007,15(6):650-655. [9] 杨月诚.焦东明,强洪夫,等.HTPB推进剂组分溶度参数的分子模拟研究[J].含能材料,2008,16(2):191-195. [10] 柯杨船,何平笙.高分子物理教程[M].北京:化学工业出版社,2006:47-52. [11] 马德柱,何平笙,徐种德.高聚物的结构与性能[M].北京:科学出版社,2004:488. [12] 吴其晔,冯莺.高分子材料概论[M].北京:机械工业出版社,2004:154-160. [13] Jawalkar S S,Aminabhavi T M.Molecular modeling simulations and thermodynamic approaches to investigate compatibility/incompatibility of poly (l-lactide)and poly (vinyl alcohol)blends[J].Polymer,2006,47(23):8061-8071. [14] Andersen H C.Molecular dynamics simulations at constant pressure and/or temperature[J].The Journal of Chemical Physics,1980,72(4):2384-2393. [15] Ewald P P.Die berechnung optischer und elektrostatischer gitterpotentiale[J].Annalen der Physik,1921,369(3):253-287. [16] Karasawa N,Goddard W A I I I.Force fields,structures,and properties of poly (vinylidene fluoride)crystals[J].Macromolecules,1992,25(26):7268-7281. [17] 付一政,刘亚青,兰艳花.端羟基聚丁二烯/增塑剂共混物相容性的分子动力学模拟[J].物理化学学报,2009,25(7):1267-1272. [18] Accelrys Corporation.Materials studio simulation tool reference manuals[M].Molecular simulation for material science,Molecular simulation in corporated:San Diego,USA;2006. (编辑:崔贤彬) Molecular simulation on selecting curing agent for NEPE propellant based on compatibility WANG Guang,HOU Shi-hai,WU Wen-ming,LI Hong-xia (No.601 Xi’an Hi-Tech Institute,Xi’an 710025,China) The molecluar models and amorphous cells of PET,N-100,TDI,and IPDI were constructed,and the solubility parameters were caculated by molecular dynamics(MD)simulation. Intermolecular pair correlation functions for pure PET,N-100,TDI, IPDI and amorphous cells of PET/N-100,PET/TDI and PET/IPDI were analyzed.Mixing energy and blends binding energy distribution of PET/N-100,PET/IPDI and PET/TDI were obtained by blending method.Different analysis comes to the same conclusion that the compatibility of PET/N-100 is better than PET/IPDI,and PET/IPDI is better than PET/TDI.The conclusion is consistent with the actual engineering application that N-100 is commonly used as curing agent in NEPE propellant,the feasibility is confirmed that we can choose curing agent based on compatibility.The method can predict compatibility of different components to promising reference for solid propellant formula design. NEPE propellant;curing agent;compatibility;molecular simulation 2014-06-26; :2014-09-18。 王广(1963—),男,教授/博导,研究方向为飞行器总体、结构分析与飞行力学。E-mail:wguang601@163.com V512 A 1006-2793(2015)05-0684-05 10.7673/j.issn.1006-2793.2015.05.0153 结论
