环丙沙星关键中间体的合成
2015-04-01管月清周国斌章鹏飞
管月清,周国斌,章鹏飞
(1.台州职业技术学院 生物与化工学院,浙江 台州 318000;2.浙江永太科技股份有限公司,浙江 临海 317016;3.台州学院 化学系,浙江 台州 318000;4.杭州师范大学 材料与化学化工学院,浙江 杭州 310036)
杂环类化合物具有不同类型的药理作用,其中氟喹诺酮类化合物具有最广谱抗菌活性,杀菌效果[1-3]。如环丙沙星、诺氟沙星、氧氟沙星、培氟沙星、依诺沙星、司帕沙星等喹诺酮类药物已在临床上被广泛应用于结膜炎和其他眼科疾病的治疗。环丙沙星[4-6]是一种氟喹诺酮类化合抗生素药物,它是第二代氟喹诺酮抗菌素,它能杀死如炭疽一类的细菌,通过干扰导致DNA 复制后反转录的酶,阻止DNA和蛋白质的合成。它不仅可用以治疗尿路感染、肠道感染、淋病等,而且可用以治疗由流感杆菌、大肠杆菌、肺炎杆菌、奇异变形杆菌、普通变形杆菌、普罗菲登菌、摩根杆菌、绿脓杆菌、阴沟肠杆菌、弗劳地枸橼杆菌、葡萄球菌属等引起的骨和关节感染、皮肤软组织感染和肺炎、败血症等。
7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸酯是合成环丙沙星的关键中间体,其结构式如下:

7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸酯的合成工艺已有许多文献报道[7-9],主要是以2,4-二氯-5-氟苯甲酰乙酰甲酸甲酯为起始原料,经缩合、取代和关环反应得以实现,但工艺过程普遍存在反应条件苛刻、反应时间长、使用危险试剂、收率低、污染大、难工业化等不足。因此,开发一种高效、易工业化合成7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸酯的新方法已成为当前研究的重点课题之一。
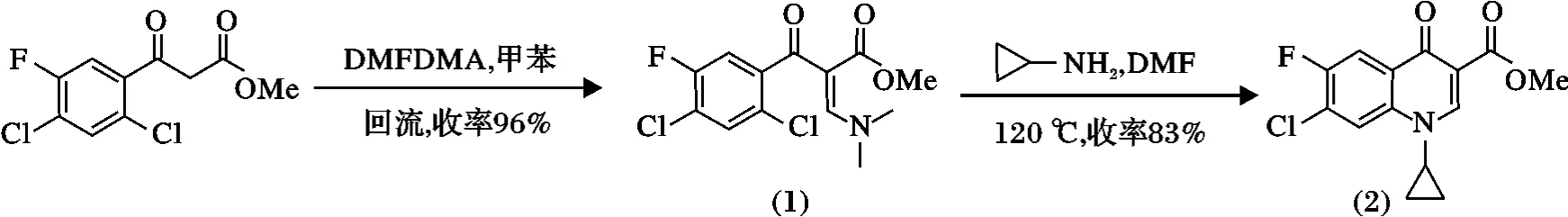
在Bunce 等[10-11]课题组研究工作的启发下,我们通过β-烯胺酮与环丙胺的串联加成消除SNAr 反应,高效地合成环丙沙星关键中间体7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸甲酯,其合成 路线如下:
1 实验部分
1.1 试剂与仪器
2,4-二氯-5-氟苯甲酰乙酸甲酯、N,N-二甲基甲酰胺二甲缩醛(DMFDMA)、环丙胺、甲苯、DMF、氯化钠、无水硫酸镁、乙酸乙酯、正己烷均为分析纯。
Mercury Plus Varian 400 型核磁共振仪;Nicolet NEXUS-470 FTIR 红外测定仪;XT-4 型显微熔点仪(温度计未校正);Thermo Scientific LCQ 质谱仪。
1.2 实验方法
1.2.1 3-二甲基氨基-2-(2,4-二氯-5-氟苯甲酰) 丙烯酸甲酯(1) 的合成 在250 mL 三颈圆底烧瓶中依次加入2,4-二氯-5-氟苯甲酰乙酰甲酸甲酯35.0 g(0.13 mol)和N,N-二甲基甲酰胺二甲缩醛(DMFDMA)的甲苯溶液(23 g DMFDMA 溶于71 mL 甲苯),加完后在回流下搅拌反应约1.5 h,TLC 跟踪,反应结束后,减压蒸馏过剩的(DMFDMA)的甲苯溶液,得40.6 g (1)油状粗产物,收率96%,直接用一下一步。分析样品为经重结晶后的白色固体,m.p.103.0 ~103.8 ℃。1H NMR(CDCl3):δ 2.97(s,3H),3.37(s,3H),3.53(s,3H),7.20(d,1H,J =6.8),7.40(d,1H,J =5.2),7.86(s,1H);13C NMR(CDCl3):δ 187.61,167.44,159.41,157.65,155.66,142.14,130.88,126.03,122.07,116.52,101.56,51.21,48.12;MS(ESI):m/z 321.2[M +H]+;IR:ν 2 933,1 695,1 625,1 562,1 465,1 425,1 382,1 367,1 296,1 244,1 199,1 122,1 093,1 058,1 012,977,970,898,883,775,725 cm-1。
1.2.2 7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸甲酯(2) 的合成 在500 mL 高压釜反应器中依次加入(1)32.0 g(0.10 mol),无水DMF 400 mL和环丙胺5.7 g(7.0 mL,0.10 mol),加完后反应器充氮密封,在120 ℃下搅拌反应约16 h,反应冷却后,将反应液倒入400 g 的冰水中,然后用300 mL的乙酸乙酯萃取,分层后,水层再用氯化钠饱和以及乙酸乙酯萃取2 次。合并有机相,有机相经饱和氯化钠溶液洗涤、无水硫酸镁干燥和减压浓缩,浓缩物经乙酸乙酯、正己烷重结晶得到24.5 g 白色固体产物(2),收率为83%,m.p.255.0 ~257.5 ℃(参考文献[10],256 ℃);1H NMR(CDCl3):δ 1.19(t,2H,J =2.0),1.37(d,2H,J=5.6),3.48(m,1H),3.91(s,3H),8.00(d,1H,J =4.8),8.12(d,1H,J =7.2),8.55(s,1H);13C NMR(CDCl3):δ 172.65,165.72,156.68,154.68,149.05,137.14,128.59,126.92,119.02,113.87,110.37,52.17,34.81,8.26;MS(ESI):m/z 296.8[M + H]+;IR:ν 2 951,1 732,1 701,1 639,1 612,1 546,1 477,1 435,1 352,1 332,1 305,1 261,1 240,1 195,1 165,1 097,1 022,898,802 cm-1。
2 结果与讨论
如合成路线所示,2,4-二氯-5-氟苯甲酰乙酰甲酸甲酯与N,N-二甲基甲酰胺二甲缩醛在回流条件下进行反应,以96%的收率得到3-二甲基氨基-2-(2,4-二氯-5-氟苯甲酰)丙烯酸甲酯(1)。接着,以DMF 为溶剂,在120 ℃条件下,3-二甲基氨基-2-(2,4-二氯-5-氟苯甲酰)丙烯酸甲酯与环丙胺顺利进行串联加成消除SNAr 反应,得到7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸甲酯(2),收率为83%。该方法与文献报道的方法相比具有反应步骤减少,反应周期缩短;反应过程操作简单,三废排放减少;反应总收率提高,生产成本降低;利于工业化生产等优点。合成产物经核磁、红外和质谱等进行结构鉴定,确认为我们的目标产物。
3 结论
本文通过β-烯胺酮与环丙胺的串联加成消除SNAr 反应的研究,在高产率获得环丙沙星关键中间体7-氯-1-环丙基-6-氟-1,4-二氢-4-氧代喹啉-3-羧酸甲酯的同时,为其建立了一种高效的合成新方法,该工艺具有步骤少、产率高、后处理操作简便,适合工业化生产等优点。另外,该方法也为喹诺酮类抗菌药物的合成提供一种新思路。
[1] Park C H,Lee J,Jung H Y,et al.Identification,biological activity,and mechanism of the anti-ischemic quinolone analog[J].Bioorganic & Medicinal Chemistry,2007,15:6517-6526.
[2] Da Silva A D,De Almeida M V,De Souza M V,et al.Biological activity and synthetic metodologies for the preparation of fluoroquinolones,a class of potent antibacterial agents[J].Current Medicinal Chemistry,2003,10:21-39.
[3] 郭强,刘秉全,郭慧元.我国氟喹诺酮类抗菌药物的研究进展[J].中国医药工业杂志,2008,39(4):297-303.
[4] Chen Y L,Fang K C,Sheu J Y.Synthesis and antibacterial evaluation of certain quinolone derivatives[J].Journal of Medicinal Chemistry,2001,44(14):2374-2377.
[5] 陈见阳,蒋敬章.3-环丙基氨基-2-(2,4-二氯-5-氟苯甲酰基)丙烯酸甲酯的合成[J].中国医药工业杂志,2000,31(11):510-511.
[6] Abaee M S,Sharill R,Borhani S,et al.Convenient one pot synthesis of some fluoroquinolones in aqueous media[J].Heterocyclic Communications,2005,11(5):415-418.
[7] Oliphant C M,Green G M.Quinolones:a comprehensive review[J].Am Fam Physician,2002,65:455-464.
[8] Patel N B,Patel A L,Chanuhan H I.Synthesis of amide derivatives of quinolone and their antimicrobial studies[J].Indian Journal of Chemistry,2007,46B:126-134.
[9] 胡艾希,游天彪,谭英,等.环丙沙星的合成工艺改进[J].合成化学,2006,14:640-642.
[10] Bunce R A,Nammalwar B.Ethyl-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylates by a tandem SNAr-additionelimination reaction[J].Journal of Heterocyclic Chemistry,2012,49:658-663.
[11]Bunce R A,Lee E J,Grant M T.Ethyl-1,4-dihydro-4-oxo-3-quinolinecarboxylates by a tandem addition-elimination-SNAr reaction[J].Journal of Heterocyclic Chemistry,2011,48:620-625.
