1,4-二取代-1,2,3-三氮唑的合成研究
2015-03-15李焕新李惠萍
沈 乐 , 李焕新 , 李惠萍
(郑州大学 化工与能源学院 , 河南 郑州 450001)
•开发与研究•
1,4-二取代-1,2,3-三氮唑的合成研究
沈乐 , 李焕新 , 李惠萍
(郑州大学 化工与能源学院 , 河南 郑州450001)
摘要:以苄基氯(1a)、4-氯苄基氯(1b)、特戊酸氯甲酯(1c)为原料,通过叠氮取代和1,3-偶极环加成反应合成了三种含羟基的三氮唑衍生物1-苄基-4-羟甲基-1H-1,2,3-三氮唑(3a)、1-(4-氯-苄基)-4-羟甲基-1H-1,2,3-三氮唑(3b)、1-(特戊酸甲酯基)-4-羟甲基-1H-1,2,3-三氮唑(3c),实验应用FT-IR、1H NMR、13C NMR对产物的结构进行了表征,结果证实了合成路线的可行性,目标产物的收率分别为85.32%、90.26%、72.62%。
关键词:三氮唑 ; 1,3-偶极环加成 ; 叠氮取代 ; 化工中间体
1,2,3-三氮唑及其衍生物是一类在工业、农业、制药以及高分子材料等领域有着广泛用途的五元氮杂环化合物[1-3]。叠氮化合物和末端炔烃之间的Huisgen 1,3-偶极环加成反应是合成1,2,3-三氮唑衍生物最重要的方法,传统的Huisgen 1,3-偶极环加成反应虽由于反应条件苛刻以及区域选择性差,生成1,4-二取代和1,5-二取代产物,分离提纯比较困难,因而限制了其在有机合成中的应用。2002年,Tornoe[4]和Rostovtsev[5]分别报道了Cu(Ⅰ)催化1,3-偶极环加成反应,Cu(Ⅰ)的存在有效地克服了上述缺点,使得该类反应的反应条件温和、区域选择性好、生成单一的1,4-二取代-1,2,3-三氮唑产物,且产率高,从此开始了此类反应的系统研究。
含醇羟基的1,2,3-三氮唑类化合物是一类重要的化工中间体,醇羟基可以被卤素等亲核物质所取代生成三氮唑酯以及多核唑类化合物[6-8]。为此,笔者以廉价易得的含氯基团化合物(1a-1c)为原料,以乙醇/水为溶剂通过叠氮取代合成化合物2a-2c,然后再以Cu(Ⅰ)为催化剂,THF/H2O作溶剂的条件下,与炔丙醇发生Huisgen1,3-偶极环加成反应生成化工中间体3a-3c,以便将其用于聚合物质子交换膜的制备,并应用FT-IR,1H NMR、13C NMR对产物结构进行了表征。合成路线如下:
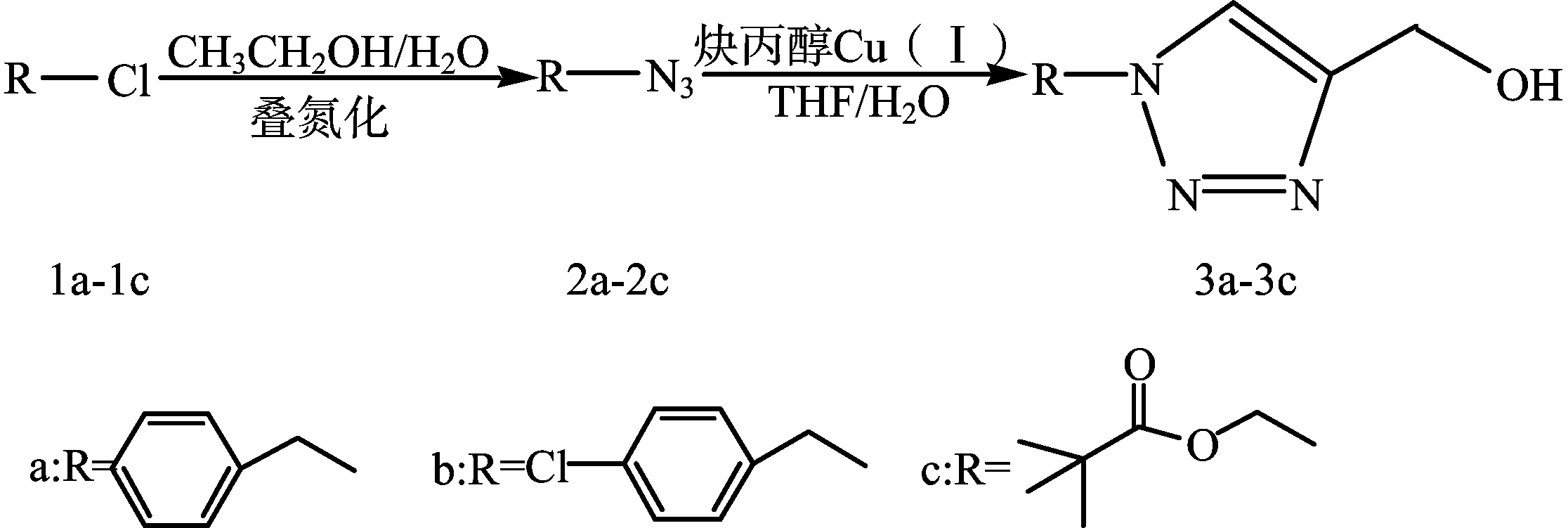
1实验部分
1.1 主要仪器和试剂
1H NMR与13C NMR表征仪器为瑞士Bruker公司DPX-400核磁共振波谱仪,以氘代氯仿为溶剂,TMS为内标。红外光谱用IR-200红外分析仪测定(KBr压片)。
二氯甲烷、无水乙醚、四氢呋喃、无水乙醇、叠氮钠、抗坏血酸、五水硫酸铜、无水硫酸钠(天津市科密欧化学试剂有限公司),苄基氯(国药集团化学试剂有限公司),4-氯苄基氯、特戊酸氯甲酯(郑州阿尔法化工有限公司),炔丙醇(郑州鑫顺电镀公司),水为实验室自制二次蒸馏水,所用药品及溶剂均为分析纯,使用前未经任何处理。
1.2 化合物的合成
1.2.12a-2c的合成(以2a为例)
在装有冷凝管和温度计的100 mL三口烧瓶中,依次加入苄基氯1a 4.8 mL,叠氮钠3.39 g,40 mL乙醇,5 mL水,在回流温度下磁力搅拌8 h。反应结束后,将反应液冷却至室温,倒入100 mL去离子水中,混合液用100 mL无水乙醚萃取两次,合并有机层,有机层经无水硫酸钠干燥24 h后过滤,滤液经减压蒸馏后得到的浅黄色液体2a为5.14 g,收率为92.10%。1H NMR(CDCl3,400 MHz),δ(×10-6):4.38(s,2H),7.39~7.45(m,3H),7.46~7.50(m,2H);13C NMR(CDCl3,100 MHz),δ(×10-6):54.83,128.31,128.38,128.92,135.50。FT-IR(KBr,cm-1):1 602~1 453,2 097。
2b、2c的合成方法如上,分别由1b以及1c与叠氮钠反应制得。
2b的产率为93.73%。1H NMR(CDCl3,400 MHz),δ(×10-6):4.33(s,2H),7.26~7.29(m,2H),7.37~7.40(m,2H);13C NMR(CDCl3,100 MHz),δ(×10-6):54.02,129.04,129.54,133.98,134.19。IR(KBr,cm-1):1 596~1 445,2 097。
2c的产率为82.29%。1H NMR(CDCl3,400 MHz),δ(ppm):1.25(s,9H),5.13(s,2H);13C NMR(CDCl3,100 MHz),δ(ppm):178.01,74.34,38.92,26.95。IR(KBr,cm-1):1 744,2 110,2 877,2 976。
1.2.23a-3c的合成(以合成3a为例)
在100 mL的三口烧瓶中依次加入苄基叠氮2a 3.33 g(25.0 mmol)、炔丙醇1.61 g(28.8 mmol)、抗坏血酸0.88 g(5.0 mmol)、CuSO4·5H2O 0.125 g(0.5 mmol)、四氢呋喃30 mL、蒸馏水10 mL,在回流温度下搅拌反应20 h,反应结束后,将反应液冷却至室温,用旋转蒸发仪除去溶剂四氢呋喃,再加入50 mL水和50 mLCH2Cl2,用分液漏斗分出有机相,水相用2×50 mL CH2Cl2萃取,合并有机层,用无水硫酸钠干燥24 h,过滤,旋蒸除去CH2Cl2,得到浅黄色固体1-苄基-4-羟甲基-1H-1,2,3-三氮唑(3a)4.40 g,产率 93.02%。
3a结构的核磁和红外表征结果:1H NMR(CDCl3,400 MHz),δ(×10-6):7.49(brs,1H),7.39~7.27(m,5H),5.51(s,2H),4.76(brs,2H),3.35(brs,—OH)。13C NMR(CDCl3,100 MHz),δ(×10-6):54.19,56.15,122.00,128.12,128.76,129.11,134.53,148.50。FT-IR(KBr,cm-1):1 013,2 097,3 261。
3b,3c的合成方法如上,分别由2b以及2c与炔丙醇反应制得。结构表征结果如下:3b的产率为96.30%。1H NMR(CDCl3,400 MHz),δ(×10-6):7.54(brs,1H),7.34~7.12(m,4H),5.47(s,2H),4.76(brs,2H),3.67(brs,-OH)。13C NMR(CDCl3,100 MHz),δ(×10-6):53.41,55.93,122.37,129.25,129.44,133.07,134.71,148.62。FT-IR(KBr,cm-1):1 012,1 659~1 464,3 257。
3c的产率为88.26%。1H NMR(CDCl3,400 MHz),δ(×10-6):7.83(s,1H),6.24(s,2H),4.80(s,2H),3.50(brs,—OH),1.18(s,9H)。13C NMR(CDCl3,100 MHz),δ(×10-6):26.79,38.80,56.08,69.73,123.32,148.30,177.80。FT-IR(KBr,cm-1):1 032,1 744,2 877,2 937,2 976,3 371。
2结果与讨论
2.1 反应条件的优化
以合成2a为例,通过单因素实验考察溶剂用量、反应温度、反应时间、反应物料物质的量比对实验收率的影响,获得较佳的反应条件。以合成3a为例,在单因素实验的基础上,通过正交试验获得合成3a的较佳反应条件。
2.2 2a合成条件的优化
2.2.1乙醇用量对产率的影响
在100 mL三口烧瓶中,依次加入苄基氯4.8 mL,叠氮钠2.90 g,5 mL蒸馏水,改变乙醇的用量,在60 ℃下反应8 h。实验结果见表1。由表1可知,随着乙醇的加入,实验收率明显增加,当乙醇用量40 mL时,产率达到最大值,而后继续增加乙醇用量,产率反而下降,这是因为溶剂量较少时不能使反应原料充分接触,而溶剂量过多时导致反应原料浓度降低,因此实验中乙醇的最佳用量为40 mL。

表1 乙醇用量对产率的影响
2.2.2反应温度对产率的影响
将溶有4.8 mL苄基氯、2.90 g叠氮钠的40 mL乙醇和5 mL水的混合溶液加入到100 mL三口烧瓶中,在电磁力搅拌下反应8 h,考察反应温度对产物收率的影响。由表2可知,随着反应温度的升高,产物收率也随着升高,并在80 ℃附近达到最大值,继续升高温度,由于乙醇/水共沸点的限制,产物收率变化不明显,故最佳反应温度为80 ℃。

表2 反应温度对产率的影响
2.2.3反应时间对产率的影响
在100 mL三口烧瓶中,依次加入苄基氯4.8 mL,叠氮钠2.90 g,40 mL乙醇和5 mL水,在磁力搅拌下回流反应,考察反应时间对产物收率的影响。实验结果如表3所示。反应时间在8 h之前时,产物收率随着反应时间增加也随之增加,但当反应时间由8 h增加到12 h,反应收率变化不大,而8 h时的收率达到最大值89.51%,由此确立最佳反应时间为8 h。

表3 反应时间对产率的影响
2.2.4反应物料物质的量比对产率的影响
在100 mL三口烧瓶中,依次加入4.8 mL苄基氯、40 mL乙醇和5 mL水,在回流温度下磁力搅拌反应8 h,改变叠氮钠的用量,考察反应物料物质的量比对产物收率的影响,见表4。

表4 物料物质的量比对产率的影响
由表4可以看出,随着反应物料物质的量比的增加,产物收率有所增加,当叠氮钠与苄基氯物质的量比为1.25∶1时产率为92.10%,此后产物收率随着叠氮钠量的增加,产率变化不明显,由此确立的最佳物料比为n(NaN3)∶n(苄基氯)=1.25∶1。
2.3 1-苄基-4-羟甲基-1H-1,2,3-三氮唑(3a)的合成
2.3.1溶剂的选择
实验选择CuSO4·5H2O和抗坏血酸原位还原产生的Cu(Ⅰ)为催化物种,由于CuSO4·5H2O和抗坏血酸在有机溶液中溶解度的原因,反应体系中要加入水作为共溶剂。目前应用较广泛的溶剂体系为叔丁醇和水,但叔丁醇可以根据底物溶解度的不同换成其它的溶剂,因此本实验选择易回收的低沸点溶剂来考察溶剂的种类对产物收率的影响。称取苄基叠氮3.33 g(0.025 mol)、炔丙醇2.8 g(0.05 mol)、抗坏血酸1.32 g(0.007 5 mol)、五水硫酸铜0.625 g(0.002 5 mol),加入蒸馏水10 mL保证CuSO4·5H2O和抗坏血酸完全溶解,加入不同的低沸点溶剂30 mL,30 ℃下反应24 h,实验结果见表5。
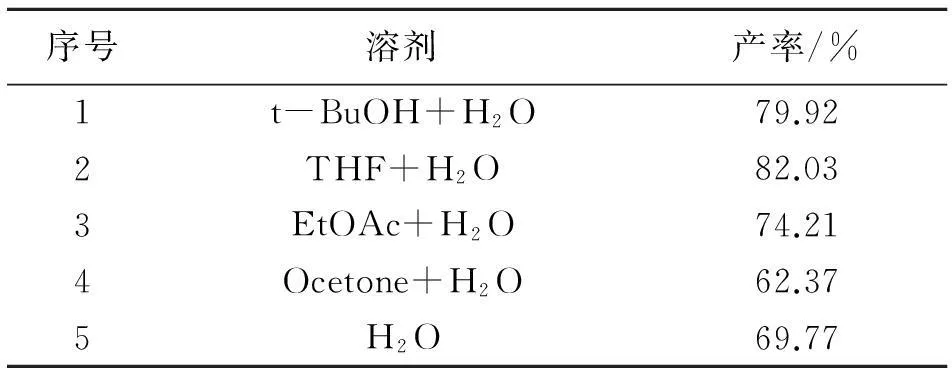
表5 不同溶剂对1-苄基-4-羟甲基-1H-1,2,3-
由表5知,当以叔丁醇和四氢呋喃与水作为共溶剂时,产率相对较高,但综合考虑成本及沸点等因素,本实验选择THF/H2O共溶体系作为反应溶剂。
2.3.2正交试验设计
通过单因素实验,初步确立了各因素的适宜范围,在此基础上选取反应温度(A),炔丙醇与苄基叠氮的物质的量比(B),反应时间(C)以及n(CuSO4·5H2O)∶n(抗坏血酸)∶n(苄基叠氮)(D)为四个影响因素,通过L9(34)正交试验确立较优的工艺条件。正交实验因素水平选择表见表6,试验方案及试验结果见表7。

表6 正交试验因素水平表

表7 正交试验方案及结果
由表7可知,因素从主到次分别为催化剂配比、反应温度、反应物物质的量比、反应时间;实验优化方案:A3B1C1D2。正交试验得出的较优实验条件为:n(炔丙醇)∶n(苄基叠氮)=1.15∶1,n(CuSO4·5H2O)∶n(抗坏血酸)∶n(苄基叠氮)=0.02∶0.20∶1,反应温度65 ℃,反应时间20 h,正交试验的9组组合中没有这组条件,按此组条件进行5次平行实验,收率分别为93.45%、93.02%、91.19%、92.18%、93.36%,平均收率为92.64%,收率虽比第7组低,但缩短了反应时间,因此确立的最佳工艺条件为:n(炔丙醇)∶n(苄基叠氮)=1.15∶1,n(CuSO4·5H2O)∶n(抗坏血酸)∶n(苄基叠氮)=0.02∶0.20∶1,反应温度65 ℃,反应时间20 h。
3结论
以含氯基团的化合物(1a-1c)和叠氮化钠为原料,通过叠氮化反应制得叠氮化合物2a-2c,反应收率分别为92.10%、93.73%、82.29%,这一方法对叠氮化合物的合成具有通用性。有机叠氮2a-2c与炔丙醇通过1,3-偶极环加成反应生成含羟基的1,2,3-三氮唑化合物3a-3c,三种目标产物的收率分别为92.64%、96.30%、88.26%。该反应属于原子经济性的绿色化学反应,反应条件温和,后处理简单。
参考文献:
[1]Yap A H,Weinreb S M.β-tosylethylazide:a useful synthon for preparation of N-protected 1,2,3-triazoles via click chemistry[J].Tetrahedron Letter,2006,47:3035-3038.
[2]Zhou Z,Li S W.Zhang Y L,et al.Promotion of proton conduction in polymer electrolyte membranes by 1H-1,2,3-triazole[J].J Am Chem Soc,2005,127(31):10824-10825.
[3]Granados-Focil S,Conway J R,Meng Y,et al.Triazole functionalized sol-gel membranes,effect of crosslink density and heterocycle content on water free proton conduction and membrane mechanical properties[J].J Macromol Sci,Pure Appl Chem,2010,47:1197-1202.
[4]Tornoe C W,Christensen C,Meldal M.Peptidotriazoles on Solid Phase:[1,2,3]-triazoles by regiospecific copper(Ⅰ)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides[J].J Org Chem,2002,67(9):3057-3064.
[5]Rostovtsev V V,Green L G,Fokin V V,et al.A stepwise huisgen cycloaddition process:copper(Ⅰ)-catalyzed regioselective“Ligation”of azides and terminal alkynes[J].Angew Chem Int Ed,2002,41,2596-2599.
[6]Maksikova A V,Sukhanov,G P,Vereshchagin L I,et al.Synthesis of triazole and tetrazole ethers[J].Izv Vyssh Uchebn Zaved,Khim Khim Tekhnol,1984,27,172-177.
[7]Vereshchagin L I,Golobokova T V,Pokatilov F A,et al.The curtius reaction in the synthesis of noncondensed poly-nuclear azoles[J].Chem Heterocycle Compd,2011,47,456-463.
[8]Golobokova T V,Pokatilov F A,Proidakov A G,et al.Synthesis of polynuclear azoles linked by ether tethers and some other products[J].Russ J Org Chem,2013,47,130-137.
Synthesis Study on of 1,4-Disubstituted-1,2,3-Triazoles
SHEN Le , LI Huanxin , LI Huiping
(School of Chemical Engineering and Energy , Zhengzhou University , Zhengzhou450001 , China)
Abstract:Using benzyl chloride(1a), 4-chloride-benzyl chloride(1b),chloromethyl pivalate(1c) as the main raw materials, three chemical intermediates 1-benzyl-4-hydroxymethyl-1H-1,2,3-triazole(3a),1-(4-chloro-benzyl) -4-hydroxymethyl-1H-1,2,3-triazole(3b), (4-(hydroxymethyl)-1,2,3-triazol-1-yl) methyl pivalate(3c) are synthesized through the azide substitution and 1,3- dipolar cycloaddition reaction.The products are characterized by FT-IR、1H NMR and13C NMR,the results confirm the feasibility of the synthetic route and the total yield of the target compounds are 85.32%、90.26%、72.62%, respectively.
Key words:1,2,3-triazole ; 1,3-dipolar cycloaddition ; azide substitution ; chemical intermediate
作者简介:沈乐(1990-),男,硕士研究生,从事有机化学合成工作;联系人:李惠萍,教授,电话:(0371)67781807。
基金项目:河南省国际合作项目(104300510009)
收稿日期:2015-03-06
中图分类号:TQ463.26
文献标识码:A
文章编号:1003-3467(2015)06-0017-04
