阿尔茨海默病转基因动物模型的学习记忆能力改变及病理学观察
2015-03-11董贤慧谢红林白江涛贺小平柴锡庆吴彦华沈瑞红
董贤慧,谢红林,白江涛,严 鹏,贺小平,柴锡庆,吴彦华,沈瑞红
阿尔茨海默病(Alzheimer’s disease,AD)是一种与年龄相关的以进行性记忆和认知功能障碍为特征的神经变性疾病。特征性病理变化表现为胆碱能神经元功能障碍、神经元和突触丢失、细胞外β-淀粉样肽(Amyloidbeta,Aβ)聚集形成老年斑(sennepfaques,SPs)和细胞内高度磷酸化的tau 蛋白形成神经原纤维缠结(neuroribrillarytangles,NFTs)。
1 材料和方法
1.1 实验动物 雄性的APPswe/PS1ΔE9双转基因小鼠与C57BL/6J 小鼠均购自北京华阜康生物科技股份有限公司。单笼喂养于光照/黑暗为12/12的恒温环境,自由摄食和饮水。
1.2 方法
1.2.1 采用Morris 水迷宫检测各组小鼠的学习记忆能力 采用Morris 水迷宫对各组小鼠进行行为认知能力测试。Morris 水迷宫主要由圆形水池和自动录像及分析系统组成。圆形水池直径120 cm、高50 cm,平台直径为10 cm,在整个实验中保持操作者位置及周围环境的相对稳定。
1.2.1.1 定位航行试验 历时5 d,每日分上、下午两个时段,固定时间为每日上午9:00~11:00,下午14:00~16:00 进行。两个时段分别从4 个不同入水点将小鼠放入水中,入水时小鼠面向水池壁,记录小鼠找到平台所用的时间,即逃避潜伏期。若小鼠入水后2 min 内未能找到平台,则将其引导置于平台上,并停留10 s,记录逃避潜伏期为120 s。每次训练时间间隔为60 s。
1.2.1.2 空间探索实验 该实验用于测量动物对平台空间位置准确记忆,即记忆保持能力。水迷宫进行6 d 将平台撤除,任选一个入水点将小鼠放入水中,游泳120 s,记录120 s 内小鼠跨过原平台的次数,即跨台次数。
1.2.2 改良Bielschowsky 银染法观察小鼠脑内老年斑 小鼠水迷宫后,甲醛灌注处死后取脑,石蜡包埋,常规石蜡切片脱腊至水,20% AgNO3水溶液37 ℃孵育30 min,4%甲醛还原,滴加银氨染液200 μl 室温孵育10 min,3 %甲醛溶液浸泡,酒精脱水、二甲苯透明、中性树脂封片,于光镜下观察。
1.2.3 尼氏染色观察 同上进行石蜡包埋、切片、脱蜡至水,置于预热至60 ℃的2 %硫瑾染色30 min后,水洗,95%酒精分色,无水酒精脱水,二甲苯透明,中性树胶封片,光镜下观察。
2 结果
2.1 Morris 水迷宫检测各组小鼠的学习记忆能力情况 水迷宫定位航行实验结果显示,APPswe/PS1ΔE9双转基因小鼠的逃避潜伏期与C57 对照组小鼠相比明显延长(P<0.05)(见图1A);空间探索实验结果显示,APPswe/PS1ΔE9双转基因小鼠与C57 对照组小鼠相比垮台次数减少(P<0.05)(见图1B)。
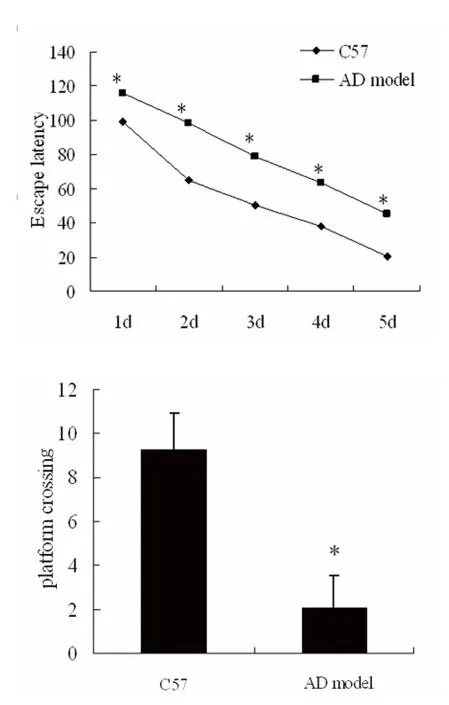
图1 Morris 水迷宫检测各组小鼠学习记忆能力情况
2.2 改良Bielschowsky 银染法观察小鼠脑内老年斑 改良Bielschowsky 银染法结果显示:C57对照组小鼠大脑皮质组织未见明显改变,神经原纤维排列整齐有序、稀疏(见图2A)。模型组小鼠大脑皮质神经原纤维肿胀,密集成宽带状,深染,可见神经纤维缠结(NFT),散在分布有老年斑(SP)(见图2B,箭头所指为老年斑)。
2.3 尼氏染色观察小鼠脑内病理改变 尼氏染色结果显示,C57 对照组小鼠海马各区神经细胞排列密集、整齐,胞浆中尼氏体丰富,大脑皮质尼氏小体呈深蓝色,细胞核淡蓝色,背景略呈浅蓝色(见图3A);APPswe/PS1ΔE9双转基因小鼠神经元水肿,细胞数量减少,排列稀疏,细胞间隙增大,胞浆内尼氏体减少,分界不清,染成淡蓝色(见图3B)。
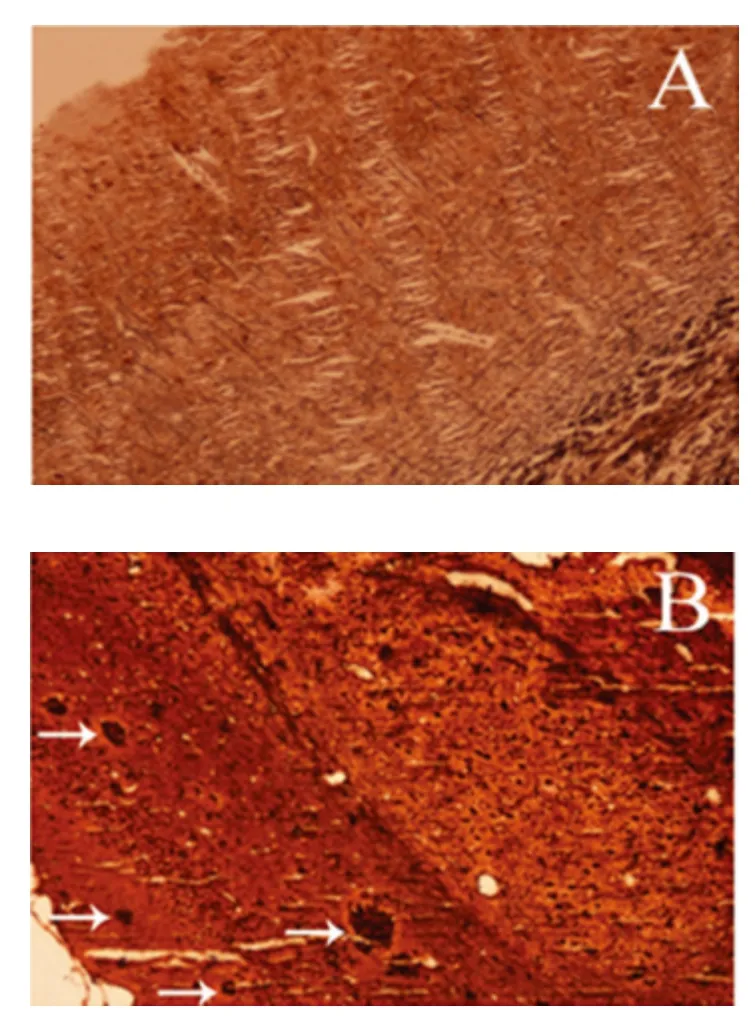
图2 改良Bielschowsky 银染法观察小鼠脑内老年斑情况

图3 尼氏染色法观察小鼠脑组织病理学变化
3 讨论
AD 是较为常见的老年人慢性退行性神经系统疾病,目前其病因及其发病机制还不是十分清楚,一定程度上制约了其治疗药物的筛选。
研究AD 有效治疗药物的主要障碍之一就是缺乏合适的AD 动物模型。建立合格的动物模型,可以在动物身上复制人类疾病模型,用于研究人类疾病的发病机制及治疗和预防措施。有关AD 动物模型,人们已经设计了很多种,包括Aβ 模型[1]、胆碱能系统损伤模型[2]、tau 蛋白模型[3]、铝模型[4]等,以上这些模型在一定程度上复制了AD 的部分特征性病理改变,但是仍然不能完全模型AD 的渐进性退行性改变,都不能算做理想的AD 动物模型。
AD 的发生是一个复杂的、多元的过程,其中遗传因素是一个重要因素。研究发现,AD 的发病与1号、14 号、19 号及21 号染色体等密切相关[5],其中包括APP 基因、Tau 蛋白基因[6]、早老蛋白(Presenilin,PS)基因[7]、UBQLN1(ubiquilin-1)蛋白基因[8]、载脂蛋白E(Apolipoprotein E,ApoE)基因[9]、α2 巨球蛋白基因(α2M)[10]、组织蛋白酶D 基因(CTSD基因)、丁酰胆碱酯酶基因(BCHE 基因)、超氧化物岐化酶基因、脂蛋白受体相关蛋白基因(LRP 基因)、α1-抗糜蛋白酶基因、血管紧张素转换酶基因(ACE 基因)、突触核蛋白基因及二氢脂酰胺琥珀酰转移酶基因以及其他许多不直接生成蛋白质的调节元素。除此以外,转基因动物(transgenic animal)技术日趋成熟,该技术可以在活体上研究某一特定致病基因的作用,这为建立AD 转基因动物模型提供了技术条件。
有关AD 转基因动物模型,其种类很多,包括单转基因动物模型,像PDAPP 小鼠[11]、Tg2576[12]、APP23[13]、PS 转基因小鼠[14]、JNPL3[15]等;双转基因动物模型,像 APPswe/PS1M146L、APPSL/PS1M146L、APPSL/PS1kI、APP KM670/671NL/APP V717F[16]、APP/ApoE 等;还包括多重转基因动物模型,如APP/PS1/tau[17,18]、Cdk5/P35/tau 等。而 目前应用最为广泛的为APPswe/PS1ΔE9双转基因小鼠模型,该模型PS1 基因E9 缺失,不是灭活作用,促使其功能加强,是国际公认的AD 转基因动物模型。
本实验研究APPswe/PS1ΔE9双转基因小鼠模型行为学及病理学变化,Morris 水迷宫实验结果显示9月龄APPswe/PS1ΔE9小鼠定位航行实验与空间探索实验结果与同月龄C57 小鼠相比有明显差异,说明9 月龄APPswe/PS1ΔE9小鼠出现明显的记忆能力缺陷,这与本实验中病理学方法,改良Bielschowsky 银染法结果发现9 月龄APPswe/PS1ΔE9小鼠大脑皮质散在分布有老年斑和尼氏染色结果显示的APPswe/PS1ΔE9双转基因小鼠神经元水肿,细胞数量减少,尼氏体减少的结果相一致。
转基因动物模型是一种基于病因的动物模型,其有着特殊的遗传学优势,使得AD 转基因动物模型堪称为研究AD 发病机制及干预措施的理想动物模型。AD 转基因动物模型的建立,有利于科研工作者深入研究AD 发病机制,并有利于进一步有针对性的研究AD 的有效治疗药物。
AD 动物模型研究的重点就是转基因动物模型,但是AD 病理过程错综复杂,因此,AD 转基因动物模型还有很大的发展空间。目前,研究发现与AD 发病有关的基因位点数量有限,还需发现新的位点,明确更多与AD 发病有关的基因位点,将有利于建立新的AD 转基因动物模型。AD 多重转基因模型日渐增多,技术逐渐完善,但是要完全攻克转基因动物模型的技术困难仍有很长的路要走,如果在转基因的基础上,再加以中枢胆碱能损害、自身免疫损伤等其他致病因素,将建立更为接近人类AD 的动物模型。
AD 相关遗传因素的新发现,将使得基因构建更加完善,AD 转基因动物模型也将进一步接近AD的全部病理特征。采用交互繁殖策略来建立多重转基因动物模型,或者进一步构建更为复杂的基因,将有助于同时表达多个AD 相关的致病基因。
本实验观察APPswe/PS1ΔE9双转基因小鼠的行为学及病理学改变,证明建立的APPswe/PS1ΔE9双转基因小鼠模型是较好的阿尔茨海默病小鼠转基因动物模型,为阿尔茨海默病发病机制研究和药物研发提供了有价值的动物模型。
综上所述,未来继续推出具备所有人AD 病理特征的理想的转基因动物模型对于开发新的治疗药物,准确的检验治疗方法和预测治疗效果是至关重要的。
[1]Prakash A,Medhi B,Chopra K.Granulocyte colony stimulating factor(GCSF)improves memory and neurobehavior in an amyloid-β induced experimental model of Alzheimer’s disease[J].Pharmacol Biochem Behav,2013,110:46-57.
[2]Kuznetsova E,Schliebs R.β-Amyloid,cholinergic transmission,and cerebrovascular system-a developmental study in amouse model of Alzheimer’s disease[J].Curr Pharm Des,2013,19(38):6749-6765.
[3]Lagoja I,Pannecouque C,Griffioen G,et al.Substituted 2-aminothiazoles are exceptional inhibitors of neuronal degeneration in tau-driven models of Alzheimer’s disease[J].Eur J Pharm Sci,2011,43:386-392.
[4]Nivsarkar M,Banerjee A.Establishing the probable mechanism of L-DOPA in Alzheimer’s disease management[J].Acta Pol Pharm,2009,66(5):483-486.
[5]Masoodi TA,Al Shammari SA,Al-Muammar MN,et al.Exploration of deleterious single nucleotide polymorphisms in late-onset Alzheimer disease susceptibility genes[J].Gene,2013,512(2):429-437.
[6]Revett TJ,Baker GB,Jhamandas J,et al.Glutamate system,amyloid β peptides and tau protein:functional interrelationships and relevance to Alzheimer disease pathology[J].J Psychiatry Neurosci,2012,37(5):110-190.
[7]Nizzari M,Thellung S,Corsaro A,et al.Neurodegeneration in Alzheimer disease:role of amyloid precursor protein and presenilin 1 intracellular signaling[J].J Toxicol,2012,18:7297.
[8]Viswanathan J,Haapasalo A,Bttcher C,et al.Alzheimer’s diseaseassociated ubiquilin-1 regulates presenilin-1 accumulation and aggresome formation[J].Traffic,2011,12(3):330-348.
[9]Reiman EM,Chen K,Liu X,et al.Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease[J].Proc Natl Acad Sci USA,2009,106(16):6820-6825.
[10]Depboylu C,Lohmüller F,Du Y,et al.Alpha2-macroglobulin,lipoprotein receptor-related protein and lipoprotein receptor-associated protein and the genetic risk for developing Alzheimer’s disease[J].Neurosci Lett,2006,400(3):187-190.
[11]Lee JE,Han PL.An update of animal models of Alzheimer disease with a reevaluation of plaque depositions[J].Exp Neurobiol,2013,22(2):84-95.
[12]Eketjäll S,Janson J,Jeppsson F,et al.AZ-4217:A High Potency BACE Inhibitor Displaying Acute Central Efficacy in Different In Vivo Models and Reduced Amyloid Deposition in Tg2576 Mice[J].J Neurosci,2013,33(24):10075-10084.
[13]Nilsen LH,Melø TM,Saether O,et al.Altered neurochemical profile in the McGill-R-Thy1-APP rat model of Alzheimer’s disease:a longitudinal in vivo 1 H MRS study[J].J Neurochem,2012,123(4):532-541.
[14]Nizzari M,Thellung S,Corsaro A,et al.Neurodegeneration in Alzheimer disease:role of amyloid precursor protein and presenilin 1 intracellular signaling[J].J Toxicol,2012,187-297.
[15]Lewis J,McGowan E,Rockwood J,et al.Neurofibrillary tangles,amyotrophy and progressive motor disturbance in mice expressing mutant(P301L)tau protein[J].Nat Genet,2000,25:402-405.
[16]McLean D,Cooke MJ,Albay R,et al.Positron emission tomography imaging of fibrillar parenchymal and vascular amyloid-β in TgCRND8 mice[J].ACS Chem Neurosci,2013,4(4):613-623.
[17]Overk CR,Perez SE,Ma C,et al.Sex steroid levels and AD-like pathology in 3xTgAD mice[J].J Neuroendocrinol,2013,25(2):131-144.
[18]Marques SC,Lemos R,Ferreiro E,et al.Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice[J].Neuroscience,2012,220:256-266.
