果核药材质量标准研究
2015-03-04史蕾李若存
陈 丹,史蕾,李 柯,李若存
(1.湘潭职业技术学院,湖南 湘潭 411102; 2.湖南省中医药研究院,湖南 长沙 410013)
果核药材质量标准研究
陈 丹1,史蕾2,李 柯2,李若存2
(1.湘潭职业技术学院,湖南 湘潭 411102; 2.湖南省中医药研究院,湖南 长沙 410013)
目的建立 果核药材的质量标准,为该药用植物资源的开发利用提供科学依据。方法采用薄层色谱(TLC)法、高效液相色谱(HPLC)法、水分测定法、灰分测定法及浸出物测定法。结果果核药材没食子酸薄层色谱中,在与对照品溶液同一位置显相同颜色的斑点。HPLC法定量分析中,没食子酸进样量线性范围为0.336~2.016 g(r=0.999 9),平均回收率为103.78%,RSD为0.78%(n=6)。结论该方法简便、准确、可靠、重复性好,可作为 果核药材的质量标准。
果核;没食子酸;薄层色谱法;高效液相色谱法;质量标准
果核为漆树科植物 果 Mangifera indica L.的果核,分布于热带与亚热带地区,我国广东、广西、海南、云南为主要产区,性味酸涩、平,功能为清热消滞,用于疝气、食滞; 果核中含大量多酚类物质,对胃肠道和呼吸道感染菌株在体内外均有较好的抗菌作用及良好的安全性,主要成分包括没食子酸、没食子乙酯、没食子甲酯、间-二没食子酸甲酯、丁二酸单甲酯和对羟基苯甲酸[1-2]。其中,没食子酸为 果核中的主要有效成分,具有抗肿瘤、抗炎、抗病毒、抗氧化和细胞毒性等作用[3-5]。 果核在内蒙古、广东、广西应用较广泛,其标准收载于《广西中药材标准》,对 果核药材的来源、性状、性味、功能与主治、用法与用量、贮藏进行了规定,但未建立定性定量指标。本试验中考察了 果核中没食子酸的薄层鉴别、含量测定及一些理化指标,制订了 果核的质量标准,为更好地开发利用其资源和临床用药提供质量控制依据。
1 仪器与试药
Series 200型高效液相色谱仪(Perkin Elmer公司);AE-240型电子分析天平;超声波清洗器(武汉恒信世纪科技有限公司);Gamag TLC Visualizer型薄层扫描仪(大连依利特分析仪器有限公司);定量毛细管(美国 Drummond公司)。 果核(广东众康中药饮片有限公司,批号为20130916;广东康美药业股份有限公司,批号为 131010141;广东茂名市俊帮中药饮片有限公司,批号为 20131101;湖南上药九旺医药有限公司,批号为20120101, 20120314,20120926),经湖南省中医药研究院李若存研究员鉴定为漆树科植物 果 Mangifera indica L.的果核;没食子酸对照品(批号为110831-200302,中国药品生物制品检定所);硅胶G(青岛海浪硅胶干燥厂);甲醇、乙腈(色谱纯,湖南汇虹试剂有限公司);其他试剂均为分析纯,水为重蒸水。
2 方法与结果
2.1 薄层色谱(TLC)鉴别
药材:取 果核药材(批号为20120101)粉末1 g,共2份,加乙醇20 mL,超声处理20 min,滤过,滤液浓缩至2 mL,作为供试品溶液。另取没食子酸对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照2010年版《中国药典(一部)》附录ⅥB薄层色谱法试验,吸取上述3种溶液各5 μL,分别点于同一硅胶G薄层板上,以二氯甲烷-乙酸乙酯-甲酸(6∶4∶1)为展开剂,展开,取出,晾干,喷以1%三氯化铁乙醇溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上分别显相同颜色的斑点[6],色谱图见图1 A。
果核不同部位:分别取 果核仁、 果核药材及 果核壳粉末(批号为20120101)各1 g,加乙醇20 mL,超声处理20 min,滤过,滤液浓缩至2 mL,作为供试品溶液。照2010年版《中国药典(一部)》附录ⅥB薄层色谱法试验,吸取上述供试品溶液及药材鉴别项下对照品溶液各5 μL,方法同药材鉴别项,结果见图1 B。
不同产地 果核:取 果核药材粉末(批号分别为20130916,131010141,20131101,20120101,20120314,20120926)各 1 g,加乙醇20 mL,超声处理20 min,滤过,滤液浓缩至2 mL,作为供试品溶液。照《中国药典(一部)》附录ⅥB薄层色谱法试验,吸取供试品溶液及药材鉴别项下对照品溶液各5 μL,方法同药材鉴别项,结果见图1 C。

图1 果核药材薄层色谱图
2.2 没食子酸含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:Ultimate LP-C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.05%磷酸溶液(3∶97);柱温:25℃;流速:1.0 mL/min;检测波长:270 nm。理论板数以没食子酸峰计应不低于5 000,在此条件下的色谱图见图2。
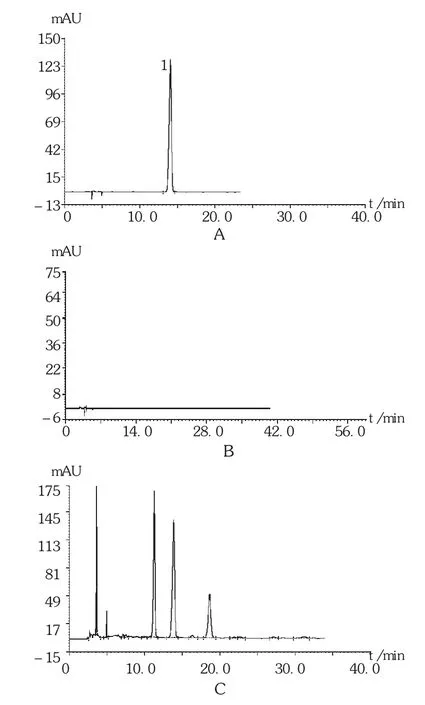
图2 果核药材高效液相色谱图
2.2.2 溶液制备
取没食子酸对照品适量,精密称定,加50%甲醇制成每1 mL含0.1 mg的溶液,即得对照品溶液。取 果核药材粉末(批号为20120101,过4号筛)约0.5 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇50 mL,密塞,称定质量,超声处理30 min,放冷,再称定质量,用50%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。以甲醇溶液为阴性对照品溶液。
在“一带一路”倡议助推我国图书出版业产业持续升级、合作模式不断创新,带动外国作家创作与讲述中国故事与中国主题并传播中国文化的过程中,外国作家及作品对“一带一路”文化进行现代阐释的积极意义不言而喻。
2.2.3 方法学考察
线性关系考察:精密吸取质量浓度为0.168 g/L的没食子酸对照品溶液2,4,6,8,10,12 μL,注入液相色谱仪,测定峰面积。以进样量(X)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,得回归方程 Y=1 772.864 0 X-10.192 8,r=0.999 9(n=6)。结果表明,没食子酸进样量在0.336~2.016 μg范围内与峰面积呈良好的线性关系。
精密度试验:精密吸取同一对照品溶液10 μL,注入液相色谱仪,连续进样6次,测定峰面积。结果的 RSD为0.42%(n=6),表明仪器精密度良好。
稳定性试验:分别精密吸取同一供试品溶液10 μL,按上述色谱条件于0,1,2,4,8,12,24 h时进样测定峰面积。结果的 RSD为1.02%(n=7),表明供试品溶液在24 h内稳定。
重复性试验:取 果核药材粉末(批号为 20120101,过4号筛)约0.5 g,共6份,精密称定,依法制备供试品溶液,精密吸取10 μL,注入液相色谱仪,测定含量。结果的 RSD为0.62%(n=6),表明方法重复性好。
加样回收试验:取没食子酸含量为13.53 mg/g的 果核药材粉末(批号为20120101,过4号筛)约 0.25 g,共 6份,精密称定,分别置具塞锥形瓶中,精密加入没食子酸对照品溶液(质量浓度为3.45 g/L)1 mL和50%甲醇49 mL,按供试品溶液的方法制备,测定含量。结果见表1。

表1 没食子酸加样回试验结果(n=6)
2.2.4 样品含量测定
2.3 不同药用部位样品测定
取 果核仁、 果核壳(批号为20120101),依法制备供试品溶液并测定含量,结果该批样品的 果核仁和 果核壳中没食子酸含量分别为1.26%和0.25%,核仁含量远高于核壳。
2.4 含量限定确定
根据4批样品含量测定结果, 果核中没食子酸含量最高1.70%,最低1.25%,平均1.48%(n=10)。考虑各种变动因素,暂订 果核中没食子酸含量不得低于1.20%。
2.5 水分、总灰分和浸出物测定
照2010年版《中国药典(一部)》附录ⅨH水分测定法(第一法烘干法)、ⅨK灰分测定法、ⅩA浸出物测定法(以水为溶剂,冷浸法)[7]依法测定。结果2批样品水分为9.9%和9.7%,总灰分为3.8%和3.9%,浸出物为18.7%和19.0%。各批次间差异不显著,故该测定结果可作为测定 果核药材质量标准的参考依据。
3 讨论
采用薄层色谱法可检出 果核药材中的主要成分没食子酸,斑点清晰且方法简便、快速、可行,故可作为 果核药材的定性鉴别依据。
取没食子酸对照品的50%甲醇溶液(0.168 g/L),经紫外扫描,在200~400 nm波长范围内记录吸收光谱。结果没食子酸在270 nm波长处有最大吸收,故采用270 nm作为检测波长。
果核占 果总重的20%~60%,其中核仁占 果核总重的45%~75%,核壳(系 果种子的坚硬外壳)占 果核总重的25% ~55%[8]。现行药材标准收载的药用部位为漆树科植物 果的果核[9]。对 果核仁和核壳含量测定结果表明,核仁中没食子酸的含量比核壳要高得多。故 果核中没食子酸含量限度的确定,是以药材标准的药用部位为依据。
[1]广西壮族自治区卫生厅.广西中药材标准[M].南宁:广西科学技术出版社,1990:55.
[2]莫武桂,刘华钢,郑陈光,等. 果核提取物动物体内外的抗菌作用及安全性研究[J].中国临床新医学,2009,2(8):779-782.
[3]谢晓艳,刘洪涛,张 吉,等.没食子酸体外抗氧化作用的研究[J].重庆医科大学学报,2011,36(3):319-322.
[4]柯发敏,张开莲.没食子酸的研究进展[J].泸州医学院学报,2011,34(4):440-442.
[5]卫智权,邓家刚,阎 莉,等. 果苷对脂多糖诱导慢性炎症的抗炎作用[J].中药药理与临床,2011,27(2):43-45.
[6]钟振国,李学坚,董明娇,等.余甘子叶中的没食子酸的定性定量分析[J].时珍国医国药,2009,20(6):1 374-1 375.
[7]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:附录52-53,附录62.
[8]Pitchaon Maisuthisakul,Michael HG.Antioxidant and tyrosinase inhibitory activity of mango seed kernel by product[J].Food Chemistry,2009,117(2):332.
[9]内蒙古自治区卫生厅.内蒙古蒙药材标准[M].赤峰:内蒙古科技出版社,1987:402.
Quality Standard of Mango Nucler L.
Chen Dan1,Shi Leizhe2,Li Ke2,Li Ruocun2
(1.Xiangtan Vocational and Technical College,Xiangtan,Hunan,China 411102; 2.Hunan Academy of TCM,Changsha,Hunan,China 410013)
Objective To provide scientific basis for the utilization and development ofMango Nucler L.by establishing its quality control standard.Methods The bioactive constituents were analyzed by TLC and HPLC.Moisture,ash and the extracts of mango nucler were all determined.Results The TLC spots of Gallic acid had similar color with the control group at the same position.The results of HPLC quantitative analysis showed that the liner range of Gallic acid was 0.336~2.016 μg(r=0.999 9),and the average recovery was 103.78%,RSD=0.78%(n=6).Conclusion This method is convenient accurate reliable with good reproducibility,so it can be used to establish quality standard for the medicinal material.
Mango Nucler L.;gallic acid;TLC;HPLC;quality standard
R284.1;R282.71
A
1006-4931(2015)21-0117-03
陈丹(1982-)女,讲师,研究方向为中药新制剂,(电子信箱)yxw0915@163.com;李柯,硕士研究生,研究方向为中药制剂与质量控制,本文通讯作者,(电话)0731-88807491(电子信箱)17848689@qq.com。
2014-08-29;
2015-05-26)
