后处理厂硝酸回收及放射性液体最小化的蒸发浓缩技术
2015-02-13刘金平叶国安
刘金平,何 辉,叶国安
中国原子能科学研究院 放射化学研究所,北京 102413
后处理厂硝酸回收及放射性液体最小化的蒸发浓缩技术
刘金平,何 辉*,叶国安
中国原子能科学研究院 放射化学研究所,北京 102413
综述了蒸发浓缩工艺在后处理厂中的应用,其主要是用于硝酸和水的循环复用及放射性废液体积的最小化。主要在以下几个方面应用:第一,后处理过程产生的1AW、2AW和2DW废液硝酸浓度均较高,这些废液的蒸发浓缩过程中,为了减小蒸发器的腐蚀,外加还原剂脱硝以控制硝酸浓度低于3mol/L、溶液温度低于100℃;第二,具有较高硝酸浓度的废液是草酸钚沉淀母液,其蒸发浓缩过程中不仅需要脱硝,还需要破坏溶液中的草酸根;第三,一些料液如1CU、2EU和2BP,硝酸浓度不高,可直接进行蒸发浓缩,无需脱硝。最后蒸发浓缩还可以处理后处理厂在启动、停车、去污、故障时产生的各种设计外的溶液。基于以上应用,蒸发浓缩技术被认为是简化和优化后处理厂的设计和操作,保证后处理厂灵活高效运行的重要技术。
蒸发浓缩;脱硝;后处理废液;草酸破坏
在后处理厂运行中,会产生含有放射性的硝酸废液和一定浓度的铀钚硝酸溶液中间产品。放射性硝酸废液,含有裂片元素、少量铀钚、次锕系元素和硝酸,通常金属离子的浓度较低,硝酸浓度则依工艺点不同,一般为3mol/L以下;铀钚硝酸溶液中间产品是硝酸铀酰或硝酸钚溶液,通常金属离子浓度为每升数十克。这些溶液均需要蒸发浓缩处理,浓缩的溶液其金属离子浓度提高,可以衔接后继的产品转化工艺或者最终废物处理工艺;馏出的稀溶液(个别伴随氮氧化物放出)与氮氧化物再生为硝酸后返回流程复用。在后处理厂氧化物固体入厂,制得固体氧化物产品出厂,硝酸溶液应在后处理过程中尽量循环使用,以降低放射性废液体积,提高后处理厂的经济性,此项技术的应用水平是衡量后处理工艺设计水平的重要指标。
在先进核能国家的后处理厂中,都非常重视蒸发浓缩技术。这是因为蒸发浓缩技术具有如下优点。
(1)循环复用硝酸和水,减少试剂消耗和废液体积。以氮氧化物-硝酸的形式循环复用元素和水,降低了试剂硝酸的消耗。
(2)去污效率高,可高效控制排放废液中放射性的总量。有效设计的蒸发浓缩装置,可易于实现去污系数达到数个量级的去污效率,蒸出液达到低放废水的水平,可后继使用或者排放;蒸残浓缩液保留近乎全部放射性,便于后继回收或者固化处理。
(3)可靠性高。蒸发浓缩对金属离子浓度和酸度在一个数量级内波动的来料溶液均能处理,是操作弹性很宽的工艺。除可以处理流程中无需严格限制浓度要求的各段产品液外,还可以处理后处理厂在启动、停车、去污、故障时产生的设计外稀溶液,这样大大地提升了后处理厂的操作弹性,降低了设计和操作难度。
例如在法国的UP3后处理厂中,就设计有6处蒸发浓缩工艺(如图1)[1],分别是高放废液的蒸发浓缩,以备高放废液中间储存后煅烧固化;一循环铀产品的级间蒸发浓缩,是将60g/L的硝酸铀酰溶液蒸发浓缩至400g/L;铀纯化循环的蒸发浓缩,将约60g/L的硝酸铀酰蒸发浓缩至1200g/L;中放废液的脱硝蒸发浓缩,主要是回收硝酸;钚纯化循环钚产品的蒸发浓缩,将钚产品液蒸发浓缩至100g/L,以便中间暂存和同位素匀化;草酸钚沉淀母液的脱硝蒸发浓缩,主要是回收硝酸的同时将钚返回。正是这些蒸发浓缩技术,保证了后处理厂的水、氮氧化物(硝酸)的循环复用,大大地降低了放射性废液的体积。蒸发浓缩是后处理厂放射性水溶液回收硝酸、回收水以备复用的重要技术。
1 蒸发浓缩在后处理中的应用
由图1可知蒸发浓缩在法国UP3后处理中有6处应用,上述6处蒸发浓缩工艺涉及8种后处理溶液,分别是1AW、1AXXW、1CU、2EU、2BP、2AW、2DW及草酸钚沉淀母液。这8种料液除了少数之间有一定的相似性外,总的来说它们组分迥异。对上述溶液进行简单的分类和介绍如下。
(1)高放废液(1AW+1AXXW)[2-4]:对燃耗为33000MWd/t(以U计)、冷却8年的乏燃料(以下讨论均以此乏燃料为对象)进行后处理,产生的高放废液其特点是裂片元素含量高(总裂片元素质量浓度为3~9g/L)、酸度高(2~3mol/L)、放射性强(其放射性活度为l09Bq/L量级、热功率为2~6W/L)、溶液氚含量高(约为0.1g/L、放射性浓度为104Bq/L)。
蒸发浓缩是高放废液管理不可缺少的步骤,高放废液应尽可能浓缩,以不出现固体沉淀为止,通常高放废液蒸发浓缩倍数为十多倍,高放浓缩液中裂变产物质量浓度可达90g/L,衔接后继储存、煅烧到固化废液量减少至不到原来的十分之一[2]。设计降低了固化前中间贮存(buffer stor-age)的体积。高放浓缩液的中间储存是必须的,因为后继的煅烧粉末化和玻璃固化操作故障率较高,有一定的中间储存能力将极大的改善后处理厂的灵活性,不至于因为玻璃固化工段发生故障而导致整个后处理厂停工。蒸出物为含氚废水、氮氧化物。氮氧化物和含氚废水复合再生为含氚硝酸,可返回溶解器和洗锝工段中用于燃料的溶解和锝的洗涤。由于脱硝引入的甲醛和洗氚工艺要使用不含氚的硝酸,使得含氚废水过量,不能全部用于生产硝酸,所以只能过量部分海洋排放。这就需要蒸发浓缩工艺的去污系数极高,可以实现从高放废液到弱放废水水平的去污。蒸发浓缩工艺和设备需要严格设计。

图1 法国UP3后处理厂的工艺概图Fig.1 Chart of the process of UP3reprocessing plant in France
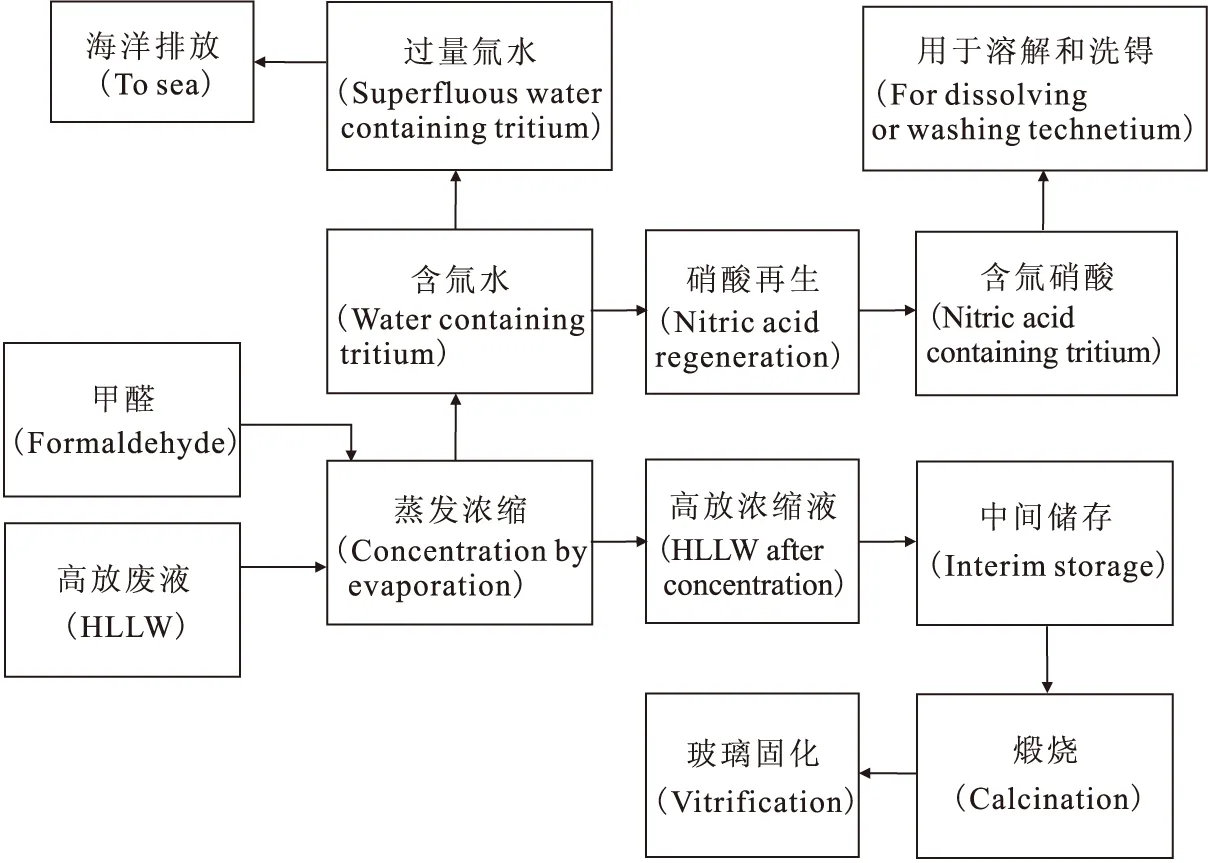
图2 高放废液处理流程图Fig.2 Chart of the discharge of HLLW
(2)中放废液(2AW+2DW):其特点是裂片元素含量很低,铀(10-2g/L)和钚(10-3g/L)浓度低,酸度高(2~3mol/L),放射性低(放射性浓度约为105Bq/L量级)。中放废液的蒸发浓缩与高放废液类似,需要甲醛脱硝,但其操作和要求相对简单,中放废液的浓缩液可以直接并入高放废液,无需再单独处置。通常中放废液蒸发浓缩倍数为80倍,因此并入高放废液的体积仅为原来的1/80。馏出的废水和氮氧化物可再生为硝酸,用于1BP(0.5mol/L)到2AF(3mol/L)、1CU(0.1mol/L)到2DF(3mol/L)的调酸。和高放废液蒸发浓缩过程一样,因为甲醛脱硝引入水而使得废水过量,过量部分作为弱放排放。这样在优化设计的后处理厂中,没有大量的中放废液需要处理,需要额外处理的中放废液仅是有机相洗涤时产生的高钠盐中放废液。
(3)铀线浓铀溶液(1CU、2EU)[2]:其特点是几乎不含裂片元素和钚,铀浓度较高(均在60g/L左右),酸度低(0.05mol/L左右)。1CU的蒸发浓缩可以提高2DF料液的U浓度,不仅减少了1CU调料过程中硝酸的加入量和2DW的废液量,1CU蒸发浓缩产生的馏出液还可以用于1CU中铀的反萃,实现废液的内循环。这就极大的降低了1CU和2DW产生的废液量。蒸发浓缩过程使得2DF的铀质量浓度达到400g/L,后继的萃取工艺因为铀的饱和度提高而对2DF残留的超铀和裂片元素等杂质去污能力大大提高。
1CU和2EU两种料液的蒸发浓缩工艺过程类似,均不涉及脱硝,两种溶液的蒸发采用两个同一类型的蒸发浓缩设备。
(4)2BP产品液[2]:2BP产品液钚浓度较高(20~40g/L左右)。严格来讲,后处理厂不同时刻产生的2BP溶液中钚的同位素组成存在差异,这会对后继MOX燃料在快堆的使用造成影响。为了解决这一问题,法国UP3后处理厂设计了2BP中间储存(buffer storage)工段,通过在特殊设计的设备中存储一段时间的钚料液使得钚同位素得到充分匀化,在2BP储存之前设计2BP蒸发浓缩,将2BP由20~40g/L浓缩到100g/L,中间储存设施设备的体积就可大大减小。
同时在后处理厂启动、停车时,需要逐渐采用硝酸置换设备里的钚,产生大量低于设计标准浓度的钚溶液,钚的浓度较低,不能直接衔接钚尾端处理。在对2BP进行蒸发浓缩时,低于设计标准浓度的钚溶液也可方便处理。这样后处理厂对于2AF浓度和2BP产品的浓度范围控制要求低,后处理厂的工艺运行难度大大降低。
(5)草酸钚沉淀母液[5-9]:草酸钚沉淀母液中钚质量浓度约为数十至100mg/L,硝酸浓度约3mol/L,草酸浓度约0.1mol/L,在一些早期的、规模较小的后处理厂中,为了消化草酸钚沉淀母液,将沉淀母液中草酸破坏后返回1BP。母液中的草酸根有可能影响2A萃取段的钚收率同时萃取过程中钚可能产生沉淀影响设备的正常运行。为了降低草酸根浓度,采用Mn2+加热破坏母液中的草酸根,但在此过程中,当草酸根浓度降低到一定程度后,草酸根再难以破坏(最大破坏率99.9%)。在此设计草酸钚沉淀母液脱硝浓缩(50~80倍),草酸的破坏量又会提高近两个数量级,这样返回到1BP中草酸根的量会降低近两个数量级,对钚纯化循环工艺的影响将大大降低。草酸钚沉淀母液的体积是2BP的10倍以上、1BP的数倍,草酸钚沉淀母液不浓缩和1BP混合会产生稀释作用,造成2AF钚浓度不高,为了解决这一问题,如法国马库尔后处理厂等,就设计了回流萃取,将一部分2BP(70%以上)、沉淀母液和1BP混合以制备2AF。
2BP和草酸钚沉淀母液的两种料液的蒸发浓缩使得后处理厂的设计和运行操作弹性加大,能耗降低,效率极大提升,工厂的运行变得更简单可靠。
由于蒸发浓缩后回收的硝酸可以在相应的后处理工艺段中复用,特别是2BP的蒸发浓缩减少了80%的钚返回钚纯化工艺段的循环,因此后处理厂中放废液分工段单独处理与集中处理相比,最终放射性废液的总量会减少,废液的单独处理还可以依据不同废液的特点,有针对性地设计蒸发浓缩工艺和设备,降低蒸发浓缩的复杂性,因此蒸发浓缩适宜在后处理厂中分工段单独应用。
综上所述,法国后处理厂中的6处蒸发浓缩工艺在乏燃料后处理中发挥着重要的作用,高放废液体积减少为原来的1/8;中放废液体积减少为原来的1/80;13%的高氚酸和约80%的低氚硝酸被回收复用;1CU和2EU分别减容6.6倍和20倍;钚产品液至少减容2.5倍,钚同位素得到匀化,草酸钚沉淀母液的处理,使返回1BP的草酸根的量减少近两个数量级,沉淀母液蒸发浓缩后不会稀释1BP;2BP无需返回2AF,2BP可以通过蒸发浓缩实现所需要的浓度,对2AF浓度、溶液酸度、流比和2BP浓度的适用范围更宽。这些优点表明,法国后处理流程的多处工段采用蒸发浓缩技术,对于流程的简化与优化、铀钚产品的减容和废液量的降低都起着非常关键的作用,蒸发浓缩技术的发展和应用会进一步的推动后处理工艺的发展,优化和完善后处理厂的操作运行。
2 后处理溶液蒸发浓缩的关键技术及国内外研究进展
上述8种溶液的蒸发浓缩涉及的关键技术主要有放射性溶液的蒸发脱硝、蒸出液的净化、草酸钚沉淀母液中草酸的破坏以及蒸发浓缩设备的设计和材料的选择。
2.1 高酸放射性溶液的蒸发脱硝
后处理厂运行产生一系列酸度较高、金属离子浓度较低的放射性废液,主要有高放废液(1AW+AXXW)、中放废液(2AW+2DW)、草酸钚盐沉淀母液,酸度均在2~3mol/L之间,金属离子质量浓度约为mg/L量级。对于溶液一般都浓缩几倍到几十倍,如不进行脱硝,硝酸浓度会一直升高,溶液的沸点也会升高(表1)。
表1 硝酸溶液沸点与硝酸浓度的关系
Table1 Relationship between the concentration of nitric acid and boiling point

w(HNO3)/%c(HNO3)/(mol·L-1)沸点(Boilingpoint)/℃w(HNO3)/%c(HNO3)/(mol·L-1)沸点(Boilingpoint)/℃00100 006013 02120 06203 54103 5668 415 16121 90305 62108 087015 70121 60407 91112 588018 44115 455010 39116 849021 17102 03
法国运行的经验表明,在硝酸浓度超过3mol/L,溶液温度超过110℃,设备腐蚀会很严重,使用寿命大大缩短,这在设备与设施要求同寿期的后处理厂中是不能接受的。所以法、日等国重点研究了高酸放射性溶液蒸发器和溶解器(工作条件相近)材料的腐蚀问题,目前锆材、钛材或者其他强化合金是研发的重点方向[10]。因此蒸发浓缩过程保持溶液中硝酸浓度不得超过3mol/L(溶液温度不超过110℃)是有严格的要求,溶液蒸发浓缩过程就必须进行脱硝。上述8种溶液,除了1CU、2EU和2BP的蒸发浓缩不需要脱硝外,其它高放废液(1AW+AXXW)、中放废物(2AW+2DW)、草酸钚盐沉淀母液的蒸发浓缩均需要脱硝。
现有后处理中脱硝工艺主要应用在高放废液和中放废液的蒸发浓缩过程中,特别是高放废液的蒸发浓缩中相关报道很多。采用的还原剂有甲醛[11-18]、甲酸[19-21]和蔗糖[22-25]。蔗糖脱硝反应易于控制、安全,但速率慢,效率依赖于高放废液中含有的Fe、Cr等[22-24],在法国UP3后处理厂中主要用于高放废液浓缩液(高浓度盐)脱硝至金属氧化物的过程。甲醛和甲酸因其脱硝速率适中、反应可控、反应产物全是气体无残渣而在脱硝中的应用最广泛,甲酸脱硝的反应速率约为甲醛的50%。
1)放射性溶液甲醛/甲酸脱硝的国内外研究现状
国外(Healy[11]、Mishra[18]等)研究了甲醛/甲酸脱硝机理,认为影响甲醛和甲酸脱硝速率的主要是酸度、温度及脱硝溶液的组成;反应存在诱导期,亚硝酸是脱硝反应的中间产物,其对脱硝反应有自催化作用,诱导期实际就是亚硝酸的积累期(亚硝酸浓度一般在10-2~10-1mol/L);诱导期也可以通过加入异相催化剂的方式缩短,如Pt/SiO2,Pd-Cu,重金属离子V、Cr、Fe、U,以及活性炭等。
德国的Wackersdorf后处理厂[3]采用甲醛连续脱硝处理高放废液,用甲酸脱硝处理中放废液。高放废液蒸发浓缩10倍,一罐3.8m3的废液需40h脱硝完毕,整个过程需要56h。回收了约13%的硝酸。中放废液蒸发浓缩20倍,尾气不回收。Wackersdorf后处理厂确定此方案时,甲醛脱硝已经在法国马库尔后处理厂得到了应用。
法国的SGN公司已经为法国的马库尔和La Hague以及日本的东海村设计了几个脱硝/浓缩设备[3],浓缩液的酸度为1~3mol/L,泡罩塔顶馏分的去污系数通常在106~108之间。通过热溶液加料(95℃)、控制料液酸度(2mol/L)和加入亚硝酸钠(100~200mg/L)来缩短反应的诱导期,保证脱硝反应的安全性。
日本原子能研究所开发了甲酸为还原剂的高放废液安全脱硝工艺[26-28]。将脱硝过程分为2个阶段:预脱硝和主要脱硝。在反应的初期控制溶液温度80℃,甲酸/硝酸摩尔比0.06~0.3,进行预脱硝。然后升高温度使溶液维持煮沸状态,连续加入甲酸脱硝。此预脱硝工艺使得反应初期安全平稳,不会产生大量气体和泡沫,有效防止馏出液的二次污染。10mol/L硝酸的脱硝反应中最大的反应热速率是209J/s,最大气体生成速率是1L/min。并研究了尿素对脱硝反应起始和终止的影响,结果表明尿素可以有效地破坏溶液中的亚硝酸,从而终止反应进行。这样,一方面可以用于终止脱硝反应,提高脱硝反应操作的安全性;另一方面也表明在脱硝反应体系中,诸如尿素、肼等能破坏亚硝酸的有机物质的存在会延长脱硝反应的诱导期,给脱硝反应增加危险。印度甘地原子研究中心[18]进行了快堆燃料后处理产生的高放废液实验室规模的甲醛脱硝实验,并依据此实验结果设计了中试规模的连续蒸发浓缩设备,并进行了相关的工艺研究。实验室规模实验中泡罩塔以φ12mm的玻璃环填充。由于残留有机溶剂的存在,反应初期会产生泡沫,但是反应过程中不需要加入抗泡沫试剂。蒸残液中剩余甲醛和甲酸的破坏有两种方式:一是蒸发浓缩停止加热后,料液加热回流1h;二是让最终得到的溶液在收集罐中室温存放24h。两种方式均可将蒸残液中的甲醛和甲酸破坏完全。金属离子和有机物的存在会使得溶液沸点升高。
美国萨瓦那河实验室研究了甲醛/甲酸脱硝过程和机理,针对亚硝酸的产生和破坏机理给出了解释[29]。2002年美国一授权专利提出了甲醛/甲酸脱硝处理放射性废液的方法[30]。采用Pt/SiO2为催化剂、控制脱硝初始反应温度为80℃、加入亚硝酸钠来保证脱硝初始反应的安全性。实验表明采用上述方法,即使在有肼和尿素等亚硝酸破坏剂存在的情况下,脱硝反应仍无诱导期,后续反应可通过控制温度和料液加入速率来保证脱硝反应的安全性。
在脱硝工艺研究中,庄维新等[31]为了分离和提取高放废液中的241Am、242Cm、147Pm、90Sr、137Cs等放射性核素,以甲酸脱硝的方法将废液的酸度从3.5mol/L降低到pH≈2,使大部分的锆、铌、铁沉淀得到去除,而镅、锔、钷、锶、铯等留于清液中。刘广成等[32]采用相同的方法研究了高放废液的脱硝条件和脱硝后锝的沉淀行为及其状态。吴传初等[33]采用此方法从高放废液中提取了铑和钯。王建晨等[34]采用甲酸脱硝研究了模拟高放废液的脱硝过程、机理以及α核素在脱硝过程中的行为,给出了各种反应条件的甲硝比。
为了用TBP萃取回收废液中的钚,中国原子能科学研究院何辉等[35]以甲酸为还原剂,对含钚0.8g/L的12.8mol/L硝酸溶液进行脱硝。实验结果表明,在冷凝回流条件下,控制体系的反应温度约为95℃,以一定的速率滴加甲酸,反应能够平稳进行,硝酸的浓度从12.8mol/L降到约为4.2mol/L,硝酸和甲酸反应的摩尔比为(1.1~1.2)∶1。
总之,国外几十年的运行经验表明,甲醛/甲酸脱硝是一种安全可控的方法,可能的安全问题主要是初始反应时诱导期导致的溶液爆沸问题和废液中有机相残留带来的“红油”问题[3,36]。
2)甲醛/甲酸脱硝反应机理
甲醛与硝酸的反应方程与硝酸浓度有关[18]:

c(HNO3)>6mol/L
(1)

c(HNO3)=2~6mol/L
(2)
c(HNO3)<2mol/L
(3)
(4)
甲酸与硝酸的反应比甲醛与硝酸的反应慢,其反应方程也与硝酸浓度有关[25]:
c(HNO3)>4mol/L
(5)
c(HNO3)<2mol/L
(6)
c(HNO3)<1mol/L
(7)
据前面介绍,在甲醛/甲酸与硝酸的反应中,亚硝酸起着非常关键的作用,下面介绍亚硝酸的生成和参与反应的机理[37]。
亚硝酸的生成反应:
(8)
(9)
(10)
(11)
亚硝酸的破坏反应:
(12)
(13)
(14)
(15)
HNO*的破坏反应:
(16)
(17)
(18)
3)影响脱硝反应的各种因素
(1)诱导期
脱硝反应在开始前会有一个诱导期的存在,从安全控制的角度考虑,这个诱导期的存在会带来较大的安全隐患,如大量反应物的聚集喷发、气压过大等。影响诱导期的参数主要有:温度、反应物浓度、亚硝酸浓度和反应器的表面特性等[38]。
HNO3浓度2~12mol/L范围内,HCOOH浓度0.2~0.4mol/L范围内,温度变化区间75~95℃下,诱导期公式如下:
当温度超过100℃时,诱导期可忽略不计。
亚硝酸浓度对诱导期有着至关重要的作用,因为亚硝酸参与反应,起到自催化作用,其极限浓度范围是10-2~10-1mol/L。通过加入亚硝酸钠可以消除诱导期,而溶液中消除亚硝酸的物质会延长诱导期,如硫酸、肼和无盐试剂等有机物。反应设备容器的比表面积越大,诱导期越短。
(2)脱硝反应的速率
脱硝速率同很多因素有关,如反应物浓度、温度、盐浓度、气压和贵金属等。
HNO3浓度0.5~4mol/L范围内,HCOOH浓度0.1~0.7mol/L范围内,脱硝反应方程如下:
HNO3浓度2~15mol/L范围内,脱硝反应方程如下[39]:
硝酸盐会加速脱硝,缩短诱导期;反应器内压力大会使NO2更好地溶解在溶液中,促进脱硝反应的进行,因此脱硝反应最好在一定的压力条件下进行;泡沫的存在有利于NO2的扩散,降低反应速率;贵金属的存在会加速反应的进行。
2.2 蒸发浓缩设备
蒸发浓缩设备中主要考虑的是蒸发浓缩过程中的加热工艺、二次冷凝液的净化和设备的腐蚀等问题。
国外的研究报告中对设备的提及很少。常用的加热器主要有外加热式蒸发器和釜式蒸发器[40]。外加热式蒸发器主要由加热室和分离室两部分组成,主要有以下两个特点:(1)加热管比较长,由于循环管未直接受到一次蒸汽加热,因此溶液的自循环速率较快,从而提高了设备的处理能力;(2)加热室和分离室分开设置,可以降低蒸发器的总高度。日本和印度的中试研究中,采用泡罩塔对蒸汽进行净化。釜式蒸发器也是由加热室和分离室两部分组成。加热器为釜型,加热部分由外加热夹套(可以设计成内部加热旋管)组成,蒸发器的顶部为分离室,内装泡罩塔和上端的水冷凝器,用于二次净化,提高去污效果。
国外釜式加热器的应用较多,最早于20世纪50年代用于法国马库尔厂,目前ARECA NC公司的阿格后处理厂的高放废液蒸发浓缩也是采用这种半连续式的釜式蒸发器[41],热交换面积仅限于蒸发器的外表面。东海村后处理厂高放废液的蒸发器同样采用釜式蒸发器,蒸发器加热系统分为外加热系统和内加热系统(由蛇形加热蒸汽管组成),塔中设置有旋风分离器和泡罩段,用以除去二次蒸汽夹带的放射性废液雾沫。此外东海村后处理厂中的酸回收蒸发器、铀浓缩蒸发器等都是采用釜式蒸发器。日本和印度的脱硝中试研究中也主要采用釜式蒸发器。釜式蒸发器有以下主要特点[18,26-28]:(1)具有一定的耐腐蚀性,蒸发器中的酸度始终保持在2~3mol/L,降低了高酸度料液对蒸发器材料的腐蚀;(2)易于实现“连续蒸发-脱硝”操作,釜式蒸发器的设计很好地解决了脱硝过程中发泡问题,通过泡罩段可以除去雾沫的夹带,降低了二次蒸汽冷凝液的放射性比活度。
制作反应器的材料主要有锆合金和不锈钢材料。法国阿格后处理厂和日本六个所村后处理厂的某些设备上采用锆702。因为锆702在强氧化介质下有很好的性能,所以在腐蚀风险较高的情况下采用这种合金。法国阿格后处理厂中放钚车间的草酸母液蒸发设备用于草酸钚母液的浓缩[41],从1983—2001年运行了18年,每年的运行时间为7680h,没有发生任何问题,每一个部件的最大厚度损失可以忽略不计。锆702的腐蚀速率非常低,设备应力腐蚀破裂的风险也非常低。日本原子力研究所研究了采用304ULC型超低不锈钢制造的减压蒸发中热交换管的腐蚀行为[42]。利用一套简化的模拟试验台架共进行了36414h的腐蚀试验,结果表明:从试验开始至超过20000h,腐蚀率增加,随后接近一个常数值(如果各项腐蚀条件和晶间腐蚀速率都无变化,那么可认定腐蚀速率保持为一常数值);从晶间腐蚀深度的测量结果看,可认为晶间腐蚀是在晶粒周围整个晶粒边界上进行的,此后晶粒脱落到溶液中;腐蚀变化的趋势主要由交换管的表面温度来确定;热交换管的液相段、沸腾发生段和气液混合段的晶间腐蚀行为没有任何差别。
法国详尽地介绍了高放废液甲醛脱硝的工艺,对此过程中的设备寿命、过程效率和化学安全问题等方面做出了总体评价[43]。阿格后处理厂40多年的运行经验表明蒸发浓缩设备的耐腐蚀性是非常好的;虽不能完全避免固体沉淀的形成,但是通过优化操作条件和工艺参数可以最大限度地降低沉淀形成的几率,再加上周期性的清洗,能使蒸发器内壁保持干净,不结垢;蒸发器的设计还能很好地解决发泡问题,确保了高且稳定的净化系数。
我国中试厂中放废液蒸发器是外加热式自然循环蒸发器,采用“清蒸”工艺,处理量低,操作运行中存在爆沸问题。胡彦涛等[44]设计了釜式蒸发器的连续蒸发-脱硝装置,并研究了反应过程中的诱导期、甲醛加入位置、温度、醛酸比、有机相和釜内压力等对硝酸破坏速率和泡沫层高度的影响,并优化了控制条件和工艺参数。并对我国后处理厂蒸发器的设计提出了建议,认为我国200t后处理厂高放废液的蒸发浓缩应采用釜式蒸发器,给出了釜式蒸发器相应的设计参数和泡罩塔设计中应考虑的问题。
2.3 草酸钚沉淀母液蒸发浓缩中草酸根的分解
对于草酸的氧化分解,有Mn(Ⅱ)催化硝酸氧化[29,45-47]、高锰酸钾催化硝酸氧化、Pt/MnO2固体催化硝酸氧化、光化学分解[48-49]、V催化硝酸氧化[50-51]、费顿化学反应[52]等方法。其中应用最广泛、成熟度最高的就是Mn(Ⅱ)催化硝酸氧化法[29]。
2009年萨瓦那河对草酸分解的方法进行了系统地评估[29]。Mn(Ⅱ)催化氧化法比其它大部分方法都快,比UV法慢,但是本方法比其它方法都有更高的成熟度。评估的目的是为了评述槽清洗液中草酸分解的方法。
Bibler等[45]总结了Mn(Ⅱ)催化硝酸氧化法的技术数据。向含有草酸的硝酸溶液中加入Mn(Ⅱ)(通常外加Mn(NO3)2),将溶液加热到100℃,体系中的硝酸会消耗草酸,反应速率可控,反应产物主要是H2O、CO2、氮氧化物(通常为NO)。Bibler报告中的草酸主要来自于槽清洗液中的草酸,清洗液中通常含有铁和铝;加入的Mn(Ⅱ)的浓度为0.01~0.02mol/L。Mn(Ⅱ)作为催化剂,可以复用,直至溶液因其它原因必须处理。Radke[53]提供了部分德国和俄罗斯关于Mn(Ⅱ)催化硝酸氧化的研究报道。其中Mirkin和Koltunov[54]研究了97℃下硝酸对草酸的氧化。他们发现硝酸浓度在1.5mol/L时有最优的反应速率,反应是自催化的,反应也存在诱导期,认为亚硝酸是促进氧化的中间产物。日本专利[55]也报道了类似的现象,专利提供了Mn(Ⅱ)催化破坏的条件,认为该过程没有爆炸危险,且反应过程可控。Gray、Burney和King[56]提出了Mn(Ⅱ)催化氧化分解硝酸用于Am的纯化。建议的流程需要溶液的蒸发,因此硝酸浓度会超过4mol/L,Mn(Ⅱ)的浓度大约是1mmol/L。分解时间和草酸分解的要求没有给出。日本专利、Namplew和Singer[57]研究了向草酸的硝酸溶液中加入甲醛/甲酸,发现草酸也会分解。认为是甲醛同硝酸的反应中间产物(HNO2)会与草酸反应。亚硝酸与草酸的反应速率远没有同甲酸快,溶液的pH值在1~2时草酸破坏速率最快,这是因为此酸度下溶液仍保持酸性且草酸可以离子化为草酸根。亚硝酸对草酸分解的促进作用在于其可以使草酸离子化。但是这些研究中没有加入Mn(Ⅱ)来研究Mn(Ⅱ)对草酸根分解的促进作用。
对于Mn(Ⅱ)破坏草酸的动力学,科学家们做了如下的研究。Bibler等[45]认为Mn(Ⅱ)催化硝酸氧化破坏草酸是一级反应,与草酸浓度和Mn(Ⅱ)浓度有关。在Mn(Ⅱ)浓度不变的情况下,可以认为该反应只与草酸浓度有关,在100℃下的速率常数为:(一级反应/min)=0.0017+0.91c(Mn2+)。该表达式表明,即使无催化剂,硝酸也会以较慢的速率分解草酸,由于铁抑制了草酸的降解,因此该式也适用于低浓度的溶液。温度对反应速率有着极大的影响,Bibler等[45]的研究给出了如下公式:
反应速率/参比速率=exp(25000/(1.987(1/K-1/Kref)))
参比温度为90℃(363.2K),由公式计算可知,当温度提高到100℃,速率提升2.6倍。这可以解释当硝酸浓度提高,溶液的沸点会升高,即使温度升高很小,反应速率也会明显增加。由上式计算出反应的活化能约为105kJ/mol。速率的控制步骤是草酸分子中的C—C键的断裂,催化物为Mn(Ⅲ)。Kubota得到的活化能为78120J/mol[45],当溶液温度低于80℃时,反应速率非常慢。
草酸根的分解在萨瓦那河也已得到了大规模的应用[29]。Bibler等[45]报道在F-Cayon蒸发器中12h内将1000磅(1磅≈0.45kg)的草酸根氧化成CO2。料液成分是0.5mol/L草酸、0.3mol/L硝酸钠和0.1mol/L的硝酸铁。蒸发罐中起初加入4mol/L的硝酸和0.02mol/L的硝酸锰。料液的加入速率略高于蒸发速率。当料液罐加满并加热到100℃后,停止加料。蒸汽的回流会将一部分的硝酸带回罐中。总的来说,消耗1mol的草酸需要2mol的硝酸。99%的草酸被破坏,反应速率常数约为0.006/min。乔继欣等[58]研究了电解氧化法分解草酸根的方法。
3 结 论
放射性液体的最小化是衡量后处理工艺设计水平的重要指标,因此法国等先进核能国家的后处理厂中都非常重视蒸发浓缩技术。后处理厂主要产生高硝酸浓度(1AW、2AW、2DW和草酸钚沉淀母液)和低硝酸浓度(1CU、2EU和2BP)的两类溶液。法国UP3后处理厂针对这些溶液的不同特点设计了6处蒸发浓缩工艺,这些工艺在后处理厂运行中起到了重要的作用,不仅大大减少了高放废液和中放废液的体积,也减少了U产品和Pu产品的体积,还能够处理后处理厂在启动、停车、去污、故障时产生的设计外稀溶液,同时还复用了大量的硝酸。蒸发浓缩技术对流程中各段产品液浓度范围要求低,能够有效地控制排放废液中放射性的总量,提高了后处理厂的操作弹性,可靠性高,降低设计和操作难度。因此蒸发浓缩技术的发展和应用在我国后处理行业应得到重视和支持。现在制约蒸发浓缩技术在后处理厂中应用的主要技术难点是:(1)能长期耐硝酸腐蚀的蒸发浓缩设备;(2)能可靠安全运行的连续蒸发浓缩工艺。今后可针对上述难点开展相关的研究工作,促进蒸发浓缩技术在我国后处理领域的应用。
[1]Debreuille M F,Vinoche R,Bailly F,et al. Reprocessing ai La Hague: industrial experience and perspectives[C]∥Global 2003,New Orleans,LA,2003: 119-123.
[2]姜圣阶,任凤仪.核燃料后处理工学[M].北京:原子能出版社,1995.
[3]Vandegrift F V. Transformation of urex effluents to solid oxides by concentration,dentration,and calcinations,ANL-00/25[R]. US: Argonne National Laboratory,2000.
[4]Cecille L,Halaszovich S. Denitration of radioactive liquid waste[G]. UK: Granam & Trotman Ltd,1986: 47-50.
[5]Schmieder H,Kroebel R. Method for preparing aqueous,radioactive waste solutions from nuclear plants for solidification: US,4056482[P]. 1977.
[6]Suzuki Y,Shimizu H,Inoue M. 1988Proceedings of the 5thinternational conference on recycling,conditioning and disposal[C]∥Recod 98. France: Conven Center-France,Vol 3,838.
[7]Lawrence F,Srinivasan R,Mathews T. Proceeding of the DAE-BRNS symposium on nuclear and radiochemistry[C]∥NUCAR 2005,Amritsar,India: 511.
[8]Mimori V,Miyajima K,Nemoto K,et al. Adsorbent of radioactive nuclides and process for volume-reduction treatment of radioactive waste: US,5476989[P]. 1995.
[9]Fanning J C. The chemical reduction of nitrate in aqueous solution[J]. Coord Chem Rev,2000,199: 159-179.
[10]王博,储凌.乏燃料后处理用设备材料Ti合金腐蚀性能研究进展[C]∥2011年后处理专业委员会年会,重庆:中核集团后处理工艺技术重点实验室,2011:136-140.
[11]Healy T V. The reaction acid with formation and with formic and its application to the removal of nitric acid from mixtures[J]. J Appl Chem,1958,8: 553-561.
[12]Kumar S V,Nadkarni M N,Myankutty T C. Chemical reduction of nitric acid is carried out by means of a homogeneous reaction with formaldehyde,Report BARC-781[R]. 1974.
[13]Evans T F. The pilot plant denitration of PUREX wastes with formaldehyde,HW-58587[R]. WA(USA): General Electric Co. Richland,1959.
[14]Morris J B. A reaction de l’acid nitrique sur le formaldehyde[J]. Energie Nucleaire,1957,1: 216.
[15]Jeffries S B. Reduction of nitrate concentration by formaldehyde treatment in the recovery of oxidation catalyst metals: US,3715320[P]. 1973.
[16]Forsman R C,Oberg G C. Formaldehyde treatment of purex radioactive wastes report,HW-79622[R]. WA(USA): General Electric Co. Richland,1963.
[17]Mishra S,Lawrence F,Srinivasan R. DAE-BRNS biennial international symposium on emerging trends in separation science and technology[C]∥SESTEC-2008,Delhi,India,2008: 195.
[18]Mishra S,Lawrence F,Sreenivasan R,et al. Development of a continuous homogeneous process for denitration by treatment with formaldehyde[J]. J Radioanal Nucl Chem,2010,285: 687-695.
[19]Kondo Y,Kubota M. Precopitation behavior of platinum group metals from simulated high liquid waste in sequential denitration process[J]. Nucl Sci Technol,1992,29: 140-148
[20]Drobnik S. Method of removing nitric acid,nitrate ions and nitrite ions out of aqueous waste solutions: US,3673086[P]. 1972.
[21]Drobnik S,Hild W,Kaufmann F,et al. Method and apparatus for processing aqueous radioactive wastes for noncontaminating and safe handling,transporting and final storage: US,4144186[P]. 1979.
[22]Bray L A. Denitration of Purex wastes with sugar,HW-76973[R]. WA(USA): General Electric Co. Richland,1963.
[23]Bray L A,Martin E C. Use of sugar to neutralize nitric acid waste liquors,HW-75565[R]. WA(USA): General Electric Co. Richland,1962.
[24]Bray L A,Martin E C. Method of treating radioactive waste: US,3158577[P]. 1964.
[25]Cecille L,Halaszovich S. Denitration of radioactive liquid waste[G]. UK: Graham & Trotman Ltd,1986: 1-10.
[26]Kondo Y. Development of a safety denitration method to remove nitric acid from miutures[J]. J Radioanal Nucl Chem,1999,240: 123-136.
[27]Kondo Y. Removal of nitric acid from a simulated high level liquid waste by a safe chemical denitration[J]. J Radioanal Nucl Chem,1999,242: 505-513.
[28]Kondo Y. Influence of urea on inition and termination of reaction between nitric acid and formic acid[J]. J Radioanal Nucl Chem,1999,242: 515-526.
[29]Nash C A. Literature review for oxalate oxidation processes and plutonium oxalate solubility,SRNL-STI-2012-00003[R]. US: Savannah River National Laboratory,2012.
[30]Broudic J C,Brossard P. Method for reducing nitrate and/or nitric acid concentration in an aqueous solution: US,6383400[P]. 2002.
[31]庄维新,邓定凯,赵沪根,等.甲酸脱硝在废水综合处理中的应用研究[J].原子能科学技术,1981(2):142-147.
[32]刘广成,江林根.从核燃料后处理废液中提取锝(Ⅰ):锝在甲酸脱硝中的沉淀行为及其状态研究[J].核科学与工程,1989,9:88-92.
[33]吴传初,刘元方,江林根.利用甲酸脱硝从核燃料后处理废液中提取贵金属铑和钯[J].核化学与放射化学,1986,8(3):147-152.
[34]王建晨,曹轩,朱永贝睿.模拟高放废液的甲酸脱硝[J].核科学与工程,1997,17(1):88-93.
[35]何辉,李斌,黄小红,等.钚萃取回收前的甲酸脱硝[M]∥中国原子能科学研究院年报.北京:原子能出版社,2007:254.
[36]Senentz G,Delvallez H. Red oil: defense in depth in the AREVA plant design[C]∥Global 2009,Paris,France,2009: 281-285.
[37]Orebaugh G E. DP-1417[R]. E I Du Pont de Nemours & Co.,1976.
[38]Cecille L,Halaszovich S. Denitration of radioactive liquid waste[G]. UK: Graham & Trotman Ltd,1986: 11-31.
[39]Healy T V,Davies B L. The destruction of nitritc acid by formaldehyde,par Ⅱ,Ⅲ and Ⅳ,AERE C/R 1739[R]. 1956.
[40]胡彦涛,杨欣静,徐云起.后处理厂高放废液釜式蒸发器的设计及问题探讨[C]∥2011年后处理专业委员会年会.重庆:中核集团后处理工艺技术重点实验室,2011.
[41]Chambrette P,Cogema C V. MA Pu oxalic mother liquor evaporator experience feedback over 18years active operating of a zirconium equipment[C]∥ Global 2005,AREVA Group,Japan P/GL0.
[42]Ueno F,Kato C,Motooka T. Intergranular corrosion mechanism of ultra-low carbon type 304stainless steel in nuclear reprocessing plant[C]∥Global 2007,Boise,Idaho: 1389-1393.
[43]Schneider J,Bretault P. Highly active liquid waste concentration using the denitration process in the french reprocessing plants[C]∥Global 2009,Paris,France,2009: 244-249.
[44]胡彦涛,李锐柔,徐聪.小装置釜式蒸发器“连续蒸发-脱硝”工艺研究[C]∥中国核学会核化工分会后处理专业委员会2013年学术交流会摘要文集.云南:中国核学会核化工分会后处理专业委员会,2013:32.
[45]Bibler N E,Hoisington J E,Holtzscheiter E W. Technical data summary destruction of oxialic acid by manganese catalyzed nitric acid reaction,DPSTD-80-36[R]. 1981.
[46]陈久宽,杨永根.对草酸与高锰酸钾反应的探究[J].实验教学与仪器,2007(7-8):63-64.
[47]王春.草酸与高锰酸钾反应的原理探究与改进建议[J].化学教育,2010(5):66-67.
[48]Martino C J. Evaluation of alternative chemical cleaning and enhanced chemical cleaning methods,SRNL-L3100-2009-00118[R]. US: Savannah River National Laboratory,2009.
[49]Jeong J,Yoon J. pH effect on OH radical production in photo/ferrioxalate systerm[J]. Water Res,2005,39(13): 2893-2900.
[50]Freidlin G N,Golubko L A,Romanova L G. Stability of oxalic acid in nitrate solutions[J]. Zhurnal Prikladnoi Khimii,1972,45(9): 2119-2121.
[51]Germain M,Pasquiou J. Process for the treatment of nitric acid aqueous effluents containing oxalic acid,usable in particular for treating the mother liquors from the precipitation of plutonium oxalate: French,FR2554434[P]. 1985.
[52]Kulik N,Panova Y,Trapido M. The fenton chemistry and its combination with coagulation for treatment of dye solutions[J]. Sep Sci Technol,2007,42: 1521-1534.
[53]Radke J H. Oxidation of oxalic and ascorbic acids by nitric acid,780000302[R]. DuPont SRP Inter Office Memorandum,1981.
[54]Mirkin I A,Koltunov V S. Kinetics of the oxidation of oxilatic acid and oxalates with aqueous nitric acid[J]. Zhurnal Fizicheskoi Khimii,1955,29: 2163-2172.
[55]Kubota M,Yamaguchi I. Decomposition of oxalic and/or nitric acid: Japanese,JP56-129079[P]. 1981.
[56]Gray L W,Burney G A,King C M. Separation of americium-241from calcium and lead using masking and multiple oxalate precipitation techniques,DP-1765[R]. August 1988.
[57]Namplew P A,Singer K. The kinetics of oxidation by nitrious and nitric acid Ⅲ: oxidation of oxalic acid by nitrous acid[J]. J Chem Soc,1956: 1143-1146.
[58]乔继欣,张虎,叶国安,等.模拟草酸钚沉淀母液中草酸电解破坏研究[J].原子能科学与技术,2008,42(11):974-979.
Technology of Concentration by Evaporation for Recycling of Nitric Acid Solution and Reduction f Radioactivity Discharge in the Reprocessing of Nuclear Fuel
LIU Jin-ping,HE Hui*,YE Guo-an
China Institute of Atomic Energy,P. O. Box 275(26),Beijing 102413,China
The application of concentration by evaporation used nuclear fuel reprocessing plant has been summarized in this article. A conclusion was drawn that concentration by evaporation is a key technology in reprocessing plant which is mainly used in recycling of nitric acid,water and reduction of radioactivity discharge. The main applications are listed as follows. The liquid releasing from extraction units,such as 1AW,2AW and 2DW,is high concentration of nitric acid during the concentration by evaporation of them so denitration with addition of reductant is necessary to remain the concentration of HNO3below 3mol/L and temperature below 100℃ and avoid corrosion of the evaporator. Another liquid of the same high concentration HNO3is the mother liquor from plutonium oxalate precipitation process and the destruction of oxalic acid should be an important issue besides the denitration during concentration by evaporation. Some liquids with low HNO3concentration,for example,1CU,2EU and 2BP,and great variety of solutions generated from the startup,shutdown and abnormal operation also need to be concentrated by evaporation. Based on the analysis,it is found that concentration by evaporation can simplify and optimize the design and operation of reprocessing plant for its effective decontamination,high operation flexibility.
concentration by evaporation; denitration; the waste from reprocessing of nuclear fuel; the destruction of oxalate
2014-04-10;
2014-06-26
刘金平(1985—),男,山东兖州人,硕士,助理研究员,分析化学专业
*通信联系人:何 辉(1973—),男,陕西咸阳人,博士,研究员,核燃料循环与材料专业
O615.1
A
0253-9950(2015)01-0001-11
10.7538/hhx.2015.37.01.0001
