6-O-取代红霉素衍生物的研究进展
2015-02-01赵娜毕小玲
赵娜,毕小玲
(中国药科大学药物化学教研室,江苏 南京 210009)
6-O-取代红霉素衍生物的研究进展
赵娜,毕小玲*
(中国药科大学药物化学教研室,江苏 南京 210009)
大环内酯类抗生素广泛用于治疗呼吸道感染,但日益泛滥的大环内酯耐药菌正影响着公众的健康。为了解决这一问题,科研工作者对大环内酯类化合物进行了大量的结构修饰。综述近年来对红霉素衍生物C6位进行的结构改造及对其生物活性的影响。
大环内酯;红霉素;6-O-取代;C6-氨基甲酸酯;6-O-烷基
红霉素(erythromycin,1)用于治疗细菌感染已有60多年了,但其在胃中酸性条件下迅速降解为无活性的产物,并导致胃肠道不良反应[1]。20世纪90年代初,第2代红霉素衍生物如罗红霉素(roxithromycin)、阿奇霉素(azithromycin)和克拉霉素(clarithromycin,2)等相继上市,药动学性质得以改善。但是,近年来随着大环内酯类耐药菌的出现,该类药物的临床疗效受到严重影响[2-3]。其中主要包括mef型耐药菌和erm型耐药菌, erm型耐药主要是A2058在erm基因编码的甲基化酶催化下发生N6,N6-二甲基化而产生耐药,mef型耐药则是由mef基因编码的能量依赖性的外排泵促使大环内酯类抗生素外排而产生耐药[4]。酮内酯作为第3代红霉素衍生物在一定程度上克服了细菌耐药问题,代表药物有泰利霉素(telithromycin,3)和喹红霉素(cethromycin,4)[5]。
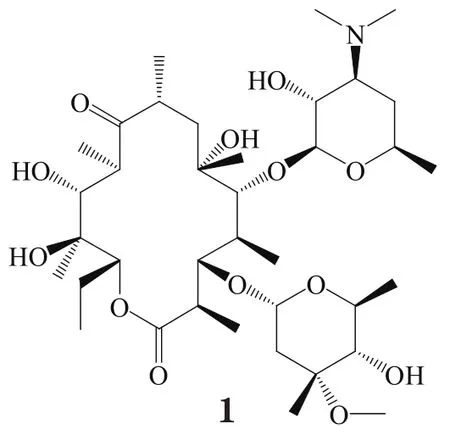

克拉霉素(2)是对红霉素(1)C6位羟基甲基化后的产物,使C9位羰基无法形成半缩酮而增加其在酸中的稳定性,具有较好的生物利用度和药动学性质,也为6-O-取代红霉素衍生物的进一步开发提供了思路[6]。研究者通过在6-OH引入烯丙基、炔丙基和氨基甲酸酯基等侧链,并在其末端引入各种芳杂环,合成了大量6-O-取代红霉素衍生物。由于大环内酯骨架上的含氧基团大多位于亲水面,C6位引入的芳杂环侧链与泰利霉素(3)中环氨基甲酸酯氮原子上连接的芳杂环侧链作用于相似的位置,即与细菌核糖体23SrRNA结构Ⅱ区的核苷酸A752结合,产生二级作用,增强抗菌活性[7-9]。
本文介绍了在不同的大环内酯母核上对C6位进行的结构修饰,尤其是结构修饰对化合物生物活性的影响,为今后该类药物的研究提供一定的参考。
1 酮内酯衍生物
研究发现,14元大环内酯C3位的L-克拉定糖可较强地诱导细菌erm基因的表达,从而导致MLSB型耐药,20世纪90年代开始兴起的酮内酯类抗生素的结构中将C3位克拉定糖替换为酮羰基使得酮内酯药物不易被“外排泵”排出,C3位酮羰基是克服细菌外排耐药机制的有效基团[10-11]。
1.1 6-OH的醚化衍生物
由雅培公司和日本大正制药联合研发的喹红霉素(4)除了将C3位改造成酮基,11、12位形成环氨基甲酸酯结构外,还将C6位的羟基醚化,即引入3-(3-喹啉基)-2(E)-丙烯基。体外抗菌试验结果显示,喹红霉素对红霉素敏感葡萄球菌的活性(MIC90=0.06 mg·L-1)是泰利霉素(MIC90=0.25 mg·L-1)的4倍,对流感嗜血杆菌的活性(MIC90=4 mg·L-1)是泰利霉素(MIC90=8 mg·L-1)的2倍。喹红霉素对肠球菌的活性(MIC90≤0.03 mg·L-1)要强于泰利霉素(MIC90=0.03 mg·L-1)[12]。
Beebe等[13]合成了一系列6-O-芳基炔丙基酮内酯化合物,该类化合物还具有9位成肟醚,11、12位为环氨基甲酸酯的结构特征。其中化合物5对ermA型耐药金葡菌的MIC为0.008 mg·L-1,活性显著强于红霉素(MIC>128 mg·L-1),是泰利霉素(MIC=0.1 mg·L-1)的12.5倍;对红霉素敏感化脓链球菌(MIC=0.008 mg·L-1)的活性强于红霉素(MIC=0.015 mg·L-1),但不及泰利霉素(MIC=0.004 mg·L-1);对ermB型耐药肺炎双球菌(MIC=0.008 mg·L-1)的活性均明显强于红霉素(MIC>128 mg·L-1)和泰利霉素(MIC=8 mg·L-1)。
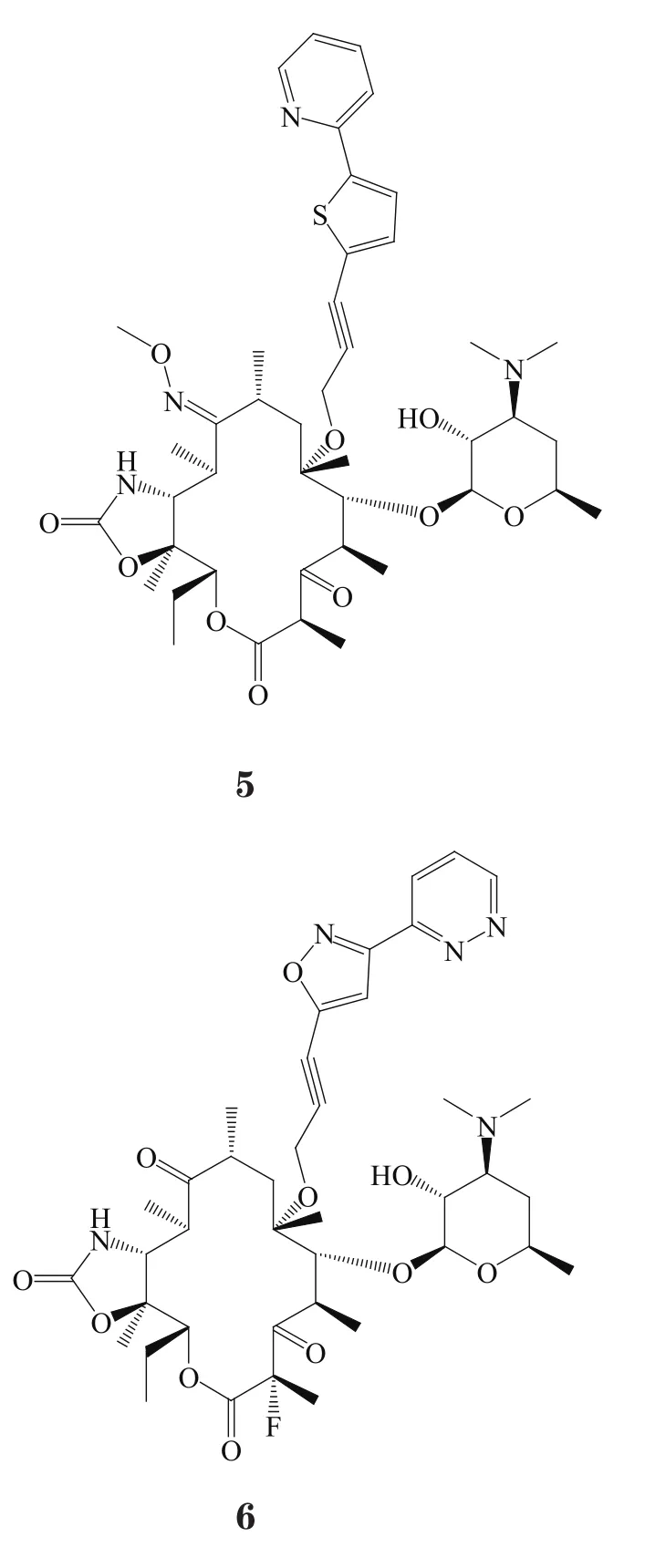
Sugimoto等[14]同样制备了6-O-芳基炔丙基-2-氟酮内酯化合物,与上述不同的是所连接芳基是异恶唑基联芳杂环。其中化合物6对红霉素敏感肺炎链球菌的活性(MIC=0.008 mg·L-1)是克拉霉素(MIC=0.03 mg·L-1)和阿奇霉素(MIC=0.06 mg·L-1)的4倍以上;对ermB型耐药肺炎链球菌的活性(MIC=0.008 mg·L-1)显著强于克拉霉素(MIC>128 mg·L-1)和阿奇霉素(MIC>128 mg·L-1);对化脓性链球菌的MIC为1 mg·L-1,而克拉霉素和泰利霉素对该细菌的MIC大于128 mg·L-1;对流感嗜血杆菌的抗菌活性(MIC=2 mg·L-1)是克拉霉素的2倍,但不如阿奇霉素(MIC=0.5 mg·L-1)。
1.2 C6-氨基甲酸酯衍生物
Henninger等[9]在C6位引入芳杂环取代的氨基甲酸酯为侧链,同时将C11、C12修饰成环氨基甲酸酯,合成了一系列化合物(7a~7h)。其中化合物7h表现出很好的抗菌活性,对各种测试菌的活性均显著强于红霉素。其对mefA型耐药肺炎链球菌、mefB型耐药肺炎链球菌的活性分别是泰利霉素的4倍和2倍;对其他测试菌的活性与泰利霉素相当(见表1)。构效关系研究表明:C6位氨基甲酸酯基侧链中氮原子与芳杂环之间以较长的丙基、丁基或烯丙基连接的化合物活性优于以甲基或乙基连接的化合物,以烯丙基最佳;所连接的芳环为含氮杂环化合物的活性好于连接苯环的化合物;连接稠合芳杂环化合物的活性强于连接非稠合双环化合物的活性。
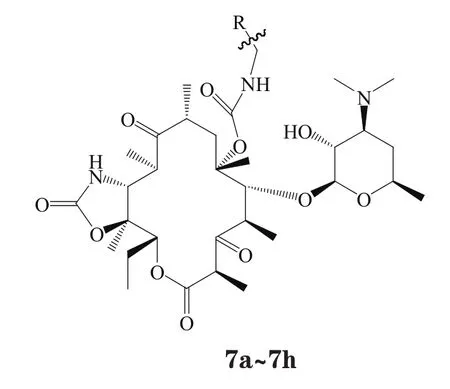
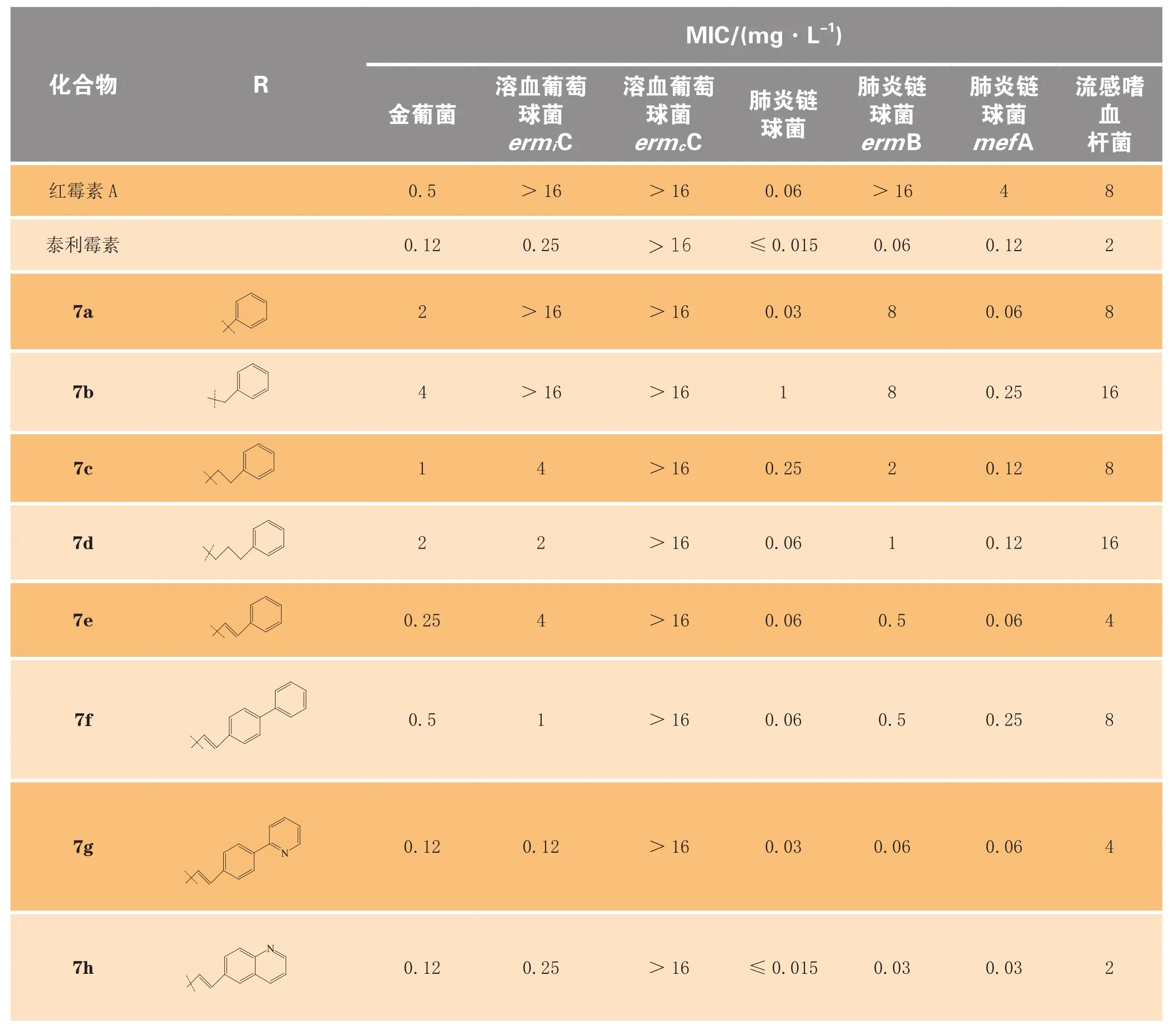
表1 C6-氨基甲酸酯酮内酯体外抗菌活性Table1 In vitro antibacterial activity of C-6 carbamate ketolides
Grant 等[15]合成了一系列C11,12-γ-内酯、C6位氨基甲酸酯化合物,其中化合物8对金葡菌的活性(MIC=0.25 mg·L-1)是红霉素(MIC=0.5 mg·L-1)的2倍;对肺炎链球菌的活性(MIC=0.03 mg·L-1)是红霉素(MIC=0.06 mg·L-1)的2倍;对ermB型耐药肺炎链球菌的活性(MIC=0.12 mg·L-1)显著强于红霉素(MIC>16 mg·L-1);对mefA型耐药肺炎链球菌的活性(MIC=0.25 mg·L-1)是红霉素(MIC=4 mg·L-1)的16倍;对流感嗜血杆菌的活性(MIC=2 mg·L-1)是红霉素(MIC=8 mg·L-1)的4倍。化合物8除对流感嗜血杆菌的活性与泰利霉素相当以外,对其他测试菌的活性均不及泰利霉素。
Zhu等[16]将C11,12-环氨基甲酸酯中的羰基O以S替换,得到一系列C11, 12-硫代环氨基甲酸酯酮内酯衍生物。其中化合物9对流感嗜血杆菌的抗菌活性(MIC=4 mg·L-1)是红霉素A(MIC=8 mg·L-1)的2倍,不及泰利霉素(MIC=2 mg·L-1);对肺炎链球菌的活性(MIC≦0.015 mg·L-1)分别是红霉素A(MIC=0.06 mg·L-1)、泰利霉素(MIC=0.03 mg·L-1)的4倍和2倍以上;对ermB型耐药肺炎链球菌、mefA型耐药肺炎链球菌的MIC分别为0.03和0.12 mg·L-1,活性分别是泰利霉素的2倍和2倍以上,均强于红霉素;对金葡菌的活性(MIC=0.25 mg·L-1)是红霉素(MIC=0.5 mg·L-1)的2倍,与泰利霉素相当。
Tennakoon等[17]合成了一系列C6-肼基甲酸酯酮内酯化合物(10a~10j),其中化合物10j对金葡菌的活性是红霉素A(MIC=1 mg·L-1)的4倍,与泰利霉素相当;对肺炎链球菌的活性均是红霉素A和泰利霉素的2倍;对流感嗜血杆菌的活性是红霉素的4倍,不及泰利霉素(见表2)。对化合物10j的体内试验表明:皮下给药时,ED50为4.8 mg·kg-1,与泰利霉素相当;而口服给药时,ED50均大于20 mg·kg-1,活性明显下降。构效关系研究表明:C6-肼基甲酸酯中β-N与芳杂环以乙基相连时活性最佳;β-N甲基化对活性影响不大,但β-N乙基化或α-N甲基化均会使MIC增加2~16倍,且2个氮原子同时甲基化会使活性明显降低。
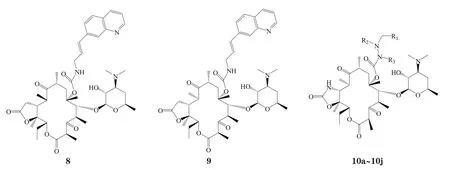
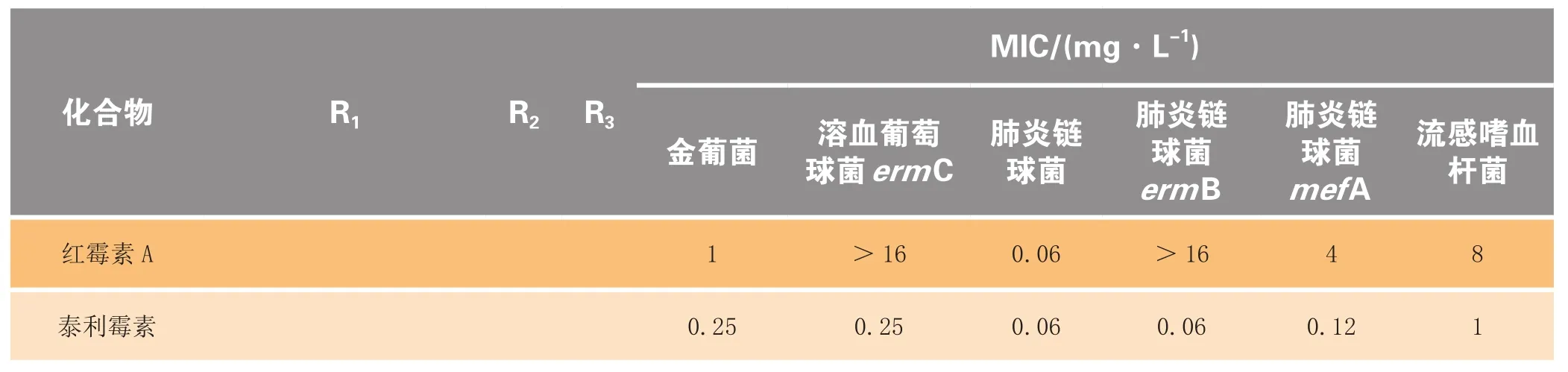
表2 C6-肼基甲酸酯酮内酯体外抗菌活性Table2 In vitro antibacterial activity of C-6 carbazate ketolides

续表2
2 酰内酯衍生物
为克服细菌的耐药性,人们对红霉素C3位克拉定糖水解后所得的羟基进行改造,除了将其转化为羰基外,还可将其酰化,从而得到另一类新型红霉素衍生物——酰内酯。
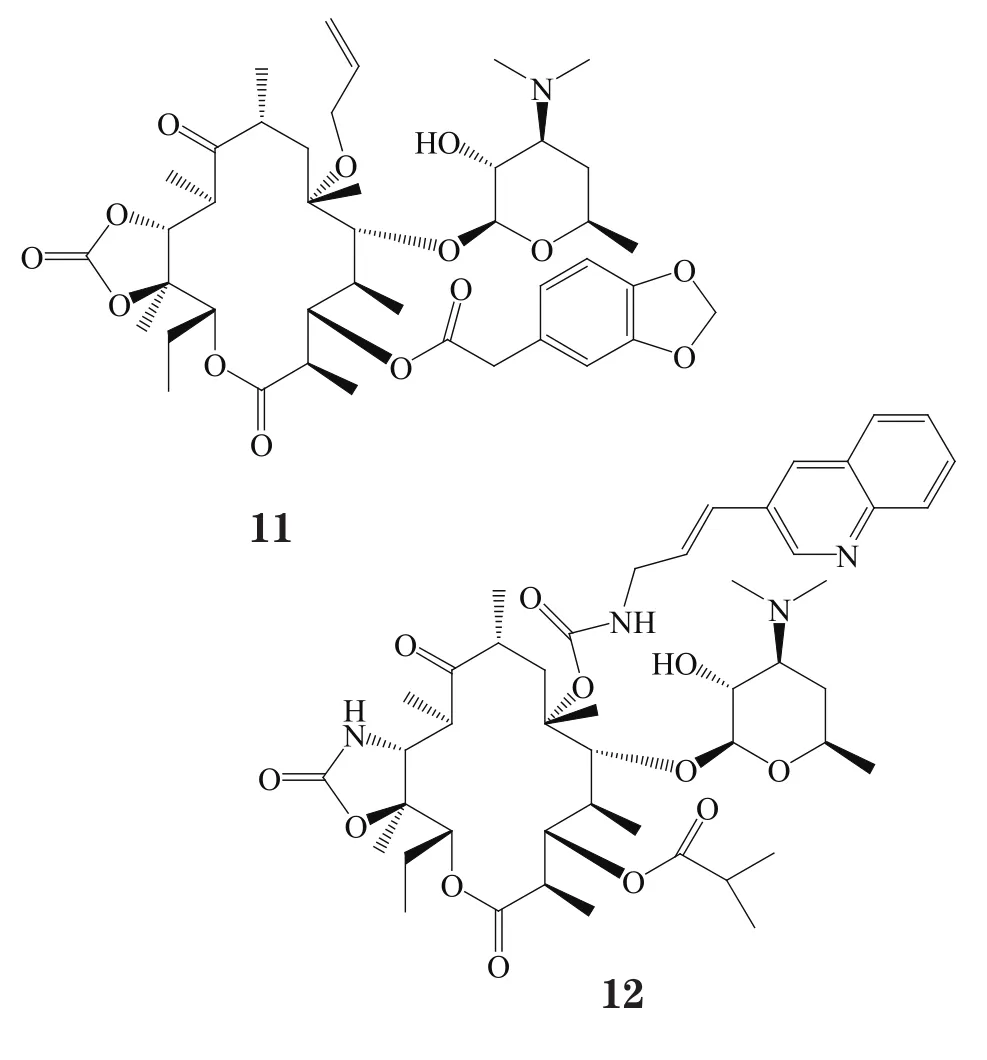
Xu等[18]将6-O-烯丙基侧链引入到酰内酯母核的C6位上,合成了一系列化合物,其中化合物11对耐甲氧西林金葡菌、表皮葡萄球菌和红霉素耐药肺炎链球菌的MIC分别为1、1和2 mg·L-1,而克拉霉素对以上细菌的MIC均为256 mg·L-1。
Zhu等[19]也将C6-氨基甲酸酯侧链引入酰内酯母环中,合成了一系列化合物。化合物12对ermB型耐药肺炎链球菌的MIC为0.06 mg·L-1,与泰利霉素相当;对mefA型耐药肺炎链球菌的活性(MIC=0.03 mg·L-1)是泰利霉素(MIC=0.25 mg·L-1)的8倍以上;对金葡菌的活性(MIC=0.25 mg·L-1)与泰利霉素相当;对流感嗜血杆菌的活性(MIC=4 mg·L-1)不及泰利霉素(MIC=2 mg·L-1)。
3 二氮内酯衍生物
在酮内酯结构的基础上,Kashimura等[20-21]将C11,12-环氨基甲酸酯酮内酯母环的C9与C11位以二氮桥相连形成七元环,得到了一类酸稳定性很强的三环酮内酯类红霉素衍生物,又称二氮内酯(diazalides)。代表化合物TE-802(13)对mef型耐药菌有很高的活性,但因结构中缺少一定长度的芳烷基侧链,故对erm型耐药菌的活性不强[8]。
Yong等[22]将芳基戊炔基侧链引入到二氮内酯的C6位的氧上,得到了化合物14,对红霉素耐药化脓性链球菌和ermB型耐药肺炎链球菌的MIC分别为0.5 和0.03 mg·L-1,活性均显著强于TE-802(MIC均大于128 mg·L-1);对红霉素耐药金葡菌的活性(MIC=0.25 mg·L-1)不如TE-802(MIC=0.1 mg·L-1);此外,C6位与芳基以戊炔基连接时的抗菌活性比以炔丙基相连时强。

4 双环内酯衍生物
大环内酯母环上有众多的活性位点和基团,将分子内不同区域的一些活性基团通过较长的链连接起来,形成跨度较大的桥环结构,所得的化合物命名为桥连双环大环内酯,简称双环内酯(bicyclolides)。
4.1 6,11-双环内酯衍生物
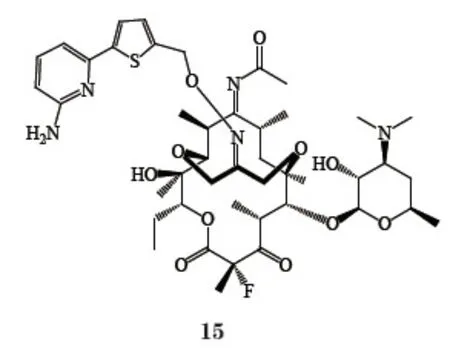
Wang等[23]开发了新型6,11-桥环酮内酯衍生物,即将C6位羟基和C11位羟基通过3个碳原子连接形成稳定的桥结构。这种桥结构能够阻止分子内缩酮化的发生,增强了化合物的稳定性,也增强了酮内酯的刚性。氧桥上通过连接不同的芳烷基侧链,与细菌核糖体产生二级作用,得到有效的抗耐药菌药物。EP-13159(15)对耐甲氧西林金葡菌的活性(MIC=4 mg·L-1)是红霉素(MIC>64 mg·L-1)的16倍以上;对流感嗜血杆菌的活性(MIC=4 mg·L-1)与红霉素相当。动物试验表
O明该化合物体内动力学性质优良,口服生物利用度可达84%,体内半衰期长达12.4 h。
4.2 3,6-双环内酯肟醚衍生物
Tang等[24]将3位和6位的羟基用2-亚甲基丙基连接起来,形成3,6-桥环,再将亚甲基氧化成羰基,与各种芳基取代的羟胺在酸催化下成肟,得到了以化合物16a为代表的一系列化合物(16a~16f,17)。化合物16a对MLSB耐药金葡菌、erm型耐药肺炎链球菌和erm型耐药化脓链球菌的活性均明显好于红霉素。研究表明:E构型肟醚(16a~16f)的活性显著大于Z构型肟醚(17);肟醚桥环上的芳杂环与肟醚氧原子直接相连所得化合物的活性最佳(见表3)。
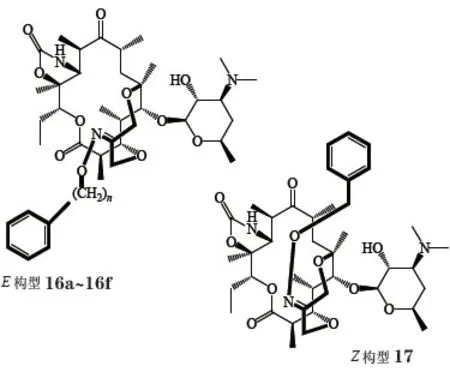
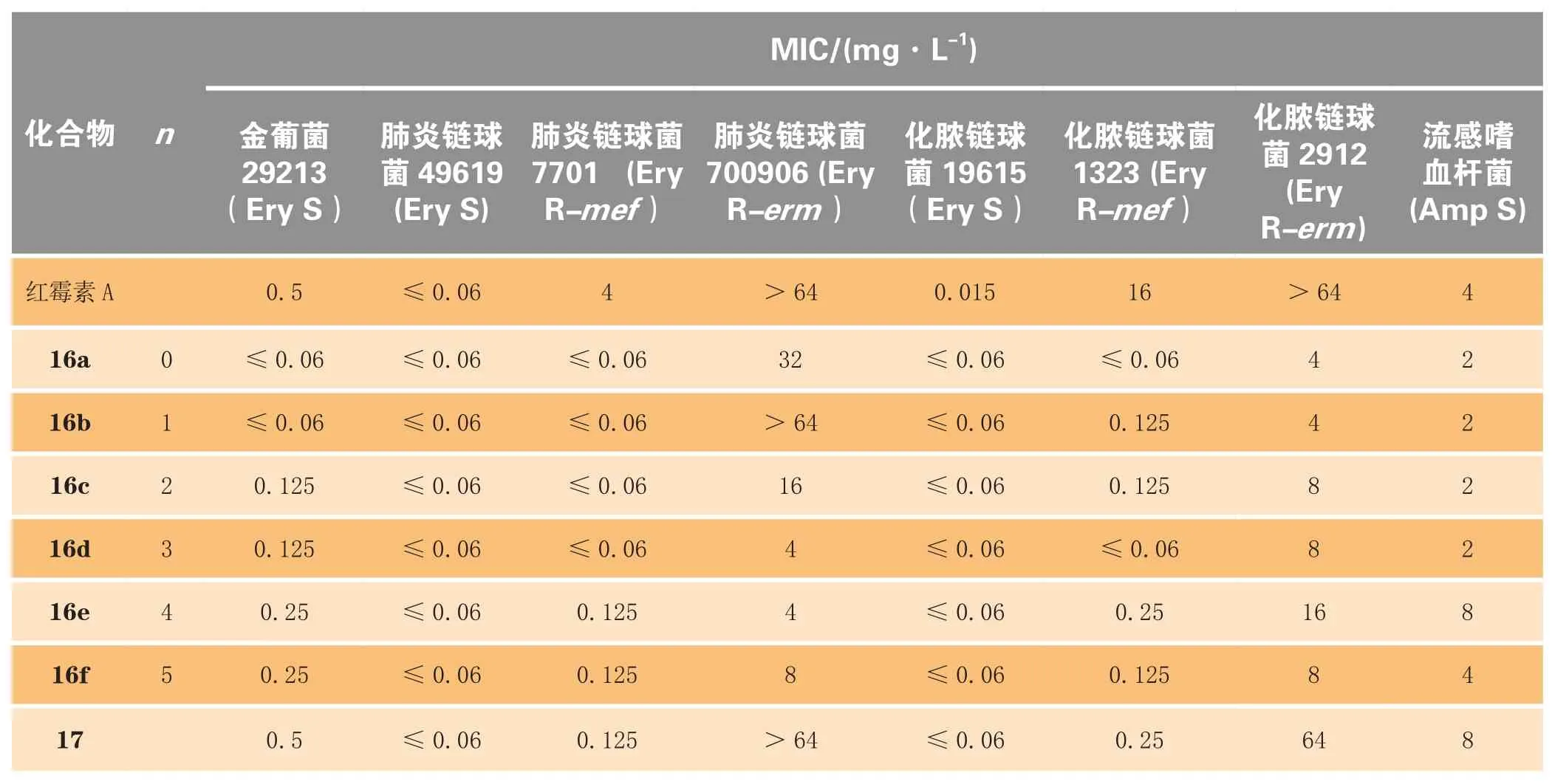
表3 3,6-双环内酯肟醚E构型化合物16a~16f和Z构型化合物17的体外抗菌活性Table3 In vitro antibacterial activities of 3,6-bicyclide E-oximes 16a-16f and Z-oxime isomer 17
5 15元氮杂内酯衍生物
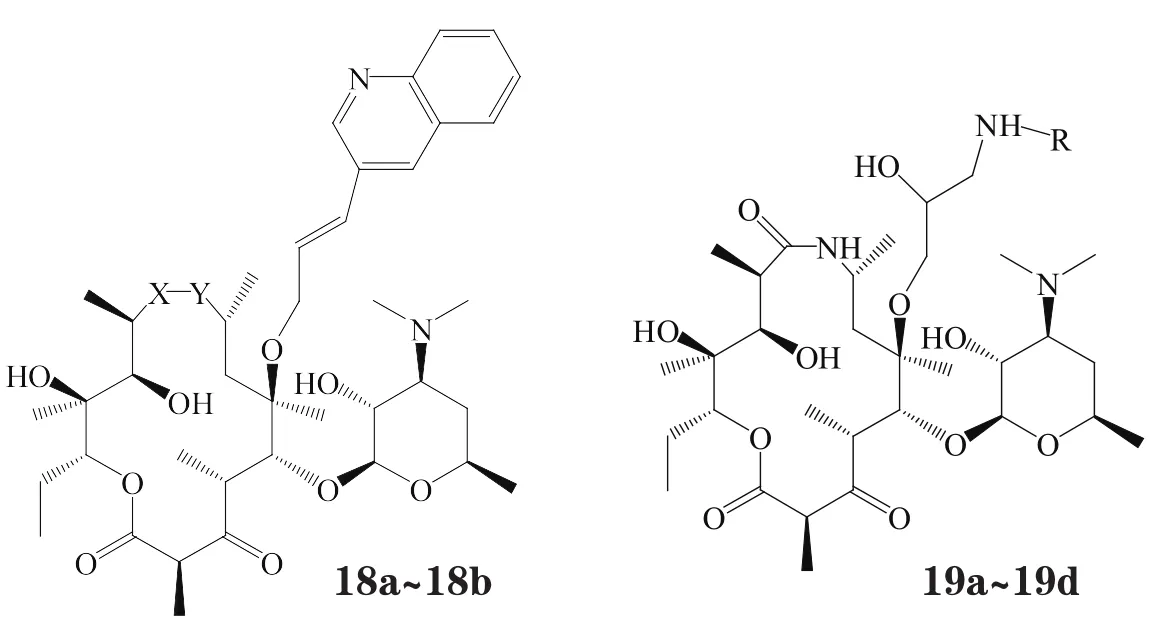
以红霉素肟(有Z、E 2种异构体)为原料,经贝克曼重排分别生成15元的8α-N和9α-N氮杂红霉素。Pavlovic'等[25]将各种芳杂环侧链引入8α-N和9α-N氮杂红霉素的C6位上,合成了一系列化合物(18a~18b;19a~19d),其中化合物19b的活性最佳,其C6位通过哌嗪链引入环丙沙星结构,形成酮内酯-喹诺酮杂合体,对部分细菌的活性优于泰利霉素、喹红霉素,表现出与环丙沙星相似的抗菌谱。构效关系研究表明:8α-N氮杂红霉素(18a)的活性强于9α-N氮杂红霉素(18b);大环内酯母环与喹诺酮环连接链的位置对活性有显著的影响,连接链在喹诺酮环C7位化合物(19b)的活性是在C6位化合物(19a)的2~32倍;以乙二胺基作为连接链的化合物(19c)的活性不如连接链是哌嗪基的化合物(19b);化合物(19d)喹诺酮环上C3位羧基被酯化后活性显著降低(见表4)。

表4 6-O-取代的8α-N和9α-N氮杂酮内酯以及酮内酯-喹诺酮杂合体的体外抗菌活性Table4 In vitro antibacterial activity of selected 6-O-substituted 8α-aza-8α-homoEry A and 9α-aza-9αhomoEry A ketolides and ketolide-quinolone hybrid
6 结语
由于大量耐药菌的出现,开发对耐药菌有效的新型大环内酯抗生素迫在眉睫。本文综述了在不同大环内酯母核上对C6位的修饰,主要包括C6-氨基甲酸酯、C6-肼基甲酸酯和6-O-烯丙基/炔丙基被各种芳杂环取代,所引入的芳杂环可以与细菌核糖体23SrRNA结构Ⅱ区的核苷酸A752结合,对部分耐药菌有一定的作用。笔者所在课题组目前正致力于将C6位与克拉定糖的4″-OH同时进行结构修饰,以期获得对耐药菌有效的红霉素衍生物。
[1]Zhang L, Chai X Y, Wang B G, et al.Design, synthesis and biological evaluation of azithromycin glycosyl derivatives as potential antibacterial agents [J].Bioorg Med Chem Lett, 2013, 23(20): 5057-5060.
[2]于丽佳,薛飞群,赵树春,等.酮内酯类抗生素研究进展[J].中兽医医药杂志,2012,31(2):24-29.
[3]Li X, Ma S T, Yan M, et al.Synthesis and antibacterial evaluation of novel 11, 4’-disubstituted azithromycin analogs with greatly improved activity against erythromycin-resistant bacteria [J].Eur J Med Chem, 2013, 59(1): 209-217.
[4]Sugimoto T, Tanikawa T, Suzuki K, et al.Synthesis and structureactivity relationship of a novel class of 15-membered macrolide antibiotics known as’11a-azalides’ [J].Bioorg Med Chem Lett, 2012, 22(20):5787-5801.
[5]Nie Y, Sun Y, You Q D.Synthesis and antibacterial activity of novel 10, 11-epoxy acylide erythromycin derivatives [J].Chin Chem Lett, 2013, 24(5):183-185.
[6]Clark R F, Ma Z H, Wang S Y, et al.Synthesis and antibacterial activity of novel 6-O-substituted erythromycin A derivatives [J].Bioorg Med Chem Lett, 2000, 10(8):815-819.
[7]Or Y S, Clark R F, Wang S Y, et al.Design, synthesis, and antimicrobial activity of 6-O-substituted ketolides active against resistant respiratory tract pathogens [J].J Med Chem, 2000, 43(10):1045-1049.
[8]Ma Z K, Clark R F, Brazzale A, et al.Novel erythromycin derivatives with aryl groups tethered to the C-6 position are potent protein synthesis inhibitors and active against multidrug-resistant respiratory pathogens [J].J Med Chem, 2001,44(24):4137-4156.
[9]Henninger T C, Xu X D, Abbanat D, et al.Synthesis and antibacterial activity of C-6 carbamate ketolides, a novel series of orally active ketolide antibiotics [J].Bioorg Med Chem Lett, 2004, 14(17): 4495-4499.
[10]孙俊,王展,张为革.抗菌活性红霉素衍生物研究进展[J].沈阳药科大学学报, 2014, 31(6): 493-504.
[11]李喆宇,崔玉彬,张静霞,等.大环内酯类抗生素的研究新进展[J].国外医药抗生素分册, 2013, 34(1):6-15.
[12]姚晓英,张永信.大环内酯类抗生素的发展和研究近况[J].上海医药, 2011, 32(7): 319-322.
[13]Beebe X, Yang F, Bui M H, et al.Synthesis and antibacterial activity of 6-O-arylpropargyl-9-oxime- 11,12-carbamate ketolides[J].Bioorg Med Chem Lett, 2004, 14(10): 2417-2421.
[14]Sugimoto T, Shimazaki Y, Manaka A, et al.Synthesis and antibacterial activity of 6-O-(heteroaryl-isoxazolyl)propynyl-2-fluoro ketolides[J].Bioorg Med Chem Lett, 2012, 22(18): 5739-5743.
[15]Grant E B, Guiadeen D, Abbanat D, et al.Synthesis and antibacterial activity of 6-O-heteroarylcarbamoyl-11, 12-lactoketolides [J].Bioorg Med Chem Lett, 2006, 16(7): 1929-1933.
[16]Zhu B, Marinelli B A, Abbanat D, et al.Synthesis and antibacterial activity of 3-keto-6-O-carbamoyl-11,12-cyclic thiocarbamate erythromycin A derivatives [J].Bioorg Med Chem Lett, 2007, 17(14):3900-3904.
[17]Tennakoon M A, Henninger T C, Darren Abbanat, et al.Synthesis and antibacterial activity of C6-carbazate ketolides[J].Bioorg Med Chem Lett, 2006, 16(24): 6231-6235.
[18]Xu P, Liu L, Jin Z P, et al.Synthesis and antibacterial activity of derivatives of 6-O-allylic acylides[J].Bioorg Med Chem Lett, 2007, 17(12):3330-3334.
[19]Zhu B, Marinelli B A, Abbanat D, et al.Synthesis and antibacterial activity of 3-O-acyl-6-O-carbamoyl erythromycin A derivatives [J].Bioorg Med Chem Lett, 2006, 16(4): 1054-1059.
[20]Kashimura M, Asaka T, Misawa Y, et al.Synthesis and antibacterial activity of the tricyclic ketolides TE-802 and its analogs[J].J Antibiot, 2001, 54(8): 664-678.
[21]Ono T, Kashimura M, Suzuki K, et al.In vitro and in vivo antibacterial activities of the tricyclic ketolide TE-802 and its analogs [J].J Antibiot, 2004, 57(8): 518-527.
[22]Yong H, Gu Y G, Clark R F, et al.Design, synthesis and structureactivity relationships of 6-O-arylpropargyl diazalides with potent activity against multidrug-resistant Streptococcus pneumoniae[J].Bioorg Med Chem Lett, 2005,15(10): 2653-2658.
[23]Wang G, Niu D, Qiu Y L, et al.Synthesis of novel 6,11-O-bridged bicyclic ketolides via a palladium-catalyzed bis-allylation[J].Org Lett, 2004, 24(6): 4455-4458.
[24]Tang D T , Gai Y H, Polemeropoulos A, et al.Design, synthesis, and antibacterial activities of novel 3,6-bicyclolide oximes: Length optimization and zero carbon linker oximes[J].Bioorg Med Chem Lett, 2008, 18(18): 5078-5082.
[25]Pavlovic´ D , Fajdetic´ A , Stjepan Mutak.Novel hybrids of 15-membered 8α- and 9α-azahomoerythromycin A ketolides and quinolones as potent antibacterials[J].Bioorg Med Chem Lett, 2010, 20(26):8566-8582.
Development in 6-O-Substituted Erythromycin Derivatives
ZHAO Na, BI Xiaoling
(Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, China)
Macrolide antibiotics are widely prescribed for the treatment of respiratory tract infections.However, the increasing prevalence of macrolide-resistant pathogens poses a serious risk to public health.To overcome the issue of drug resistance, numerous modifcations have been made on macrolides.The recent development in structural modifcations on C6 of erythromycin derivatives and the impact to biological activity was reviewed.
macrolide; erythromycin; 6-O-substituted; C6-carbamate; 6-O-alkyl
R978.1
A
1001-5094(2015)03-0179-09
接受日期:2015-01-24
*通讯作者:毕小玲,副教授;
研究方向: 半合成抗生素;
Tel:025-83271414;E-mail:bxlyy@sina.com
book=187,ebook=30
·生物制药论坛·
BIOPHARMACEUTICAL FORUM
