小分子c-Met激酶抑制剂的研究进展
2015-02-01胡亚宁胡诗合袁浩亮庄津陈亚东陆涛
胡亚宁,胡诗合,袁浩亮,庄津,陈亚东,陆涛*
(1.中国药科大学有机化学教研室,江苏 南京 211198;2.中国药科大学无机化学教研室,江苏 南京211198)
小分子c-Met激酶抑制剂的研究进展
胡亚宁1,胡诗合1,袁浩亮1,庄津1,陈亚东2,陆涛1*
(1.中国药科大学有机化学教研室,江苏 南京 211198;2.中国药科大学无机化学教研室,江苏 南京211198)
受体酪氨酸激酶c-Met即肝细胞生长因子HGF受体。HGF/c-Met信号通路在肿瘤形成、生长和转移过程中被频繁激活,因此, c-Met已成为抗癌药物研究中一个重要靶标。重点介绍近年来基于c-Met通路的抗癌药物研究进展。
c-Met激酶;HGF/c-Met通路;c-Met抑制剂;肿瘤
c-Met是肝细胞生长因子(HGF)的高亲和性受体,HGF/c-Met信号通路在伤口愈合和组织再生过程中起着重要的调节作用,并在多种恶性肿瘤中过度表达,与肿瘤的生长、转移密切相关。
c-Met是一类酪氨酸激酶跨膜糖蛋白,以通过二硫键连接而成的α-β异二聚体形式存在,α链(50 kDa)在胞外区,β链(145 kDa)包含了胞外区、跨膜区和胞内的酪氨酸激酶结构域。HGF与c-Met结合后,HGF/c-Met信号通路即被激活,首先c-Met靠近胞内区的4个磷酸化位点的酪氨酸残基发生自身磷酸化,募集下游的Gab-1、Grb-2、Shc和c-Cb1等衔接蛋白,接着通过一系列的磷酸化反应活化PI-3K、ERK1/2、PLC-γ、STAT和FAK等重要的信号分子及相应的信号通路,从而调节肿瘤细胞的增殖、迁移和侵袭能力(见图1)[1]。
c-Met的晶体结构与已知的其他蛋白激酶的结构基本一致,不同之处在于其活化环的抑制构象较为独特,与SH2结合的基序1349YVHV呈伸展构象,与GRB2-SH2结合的基序1356YVNV呈Ⅱ型β转角,中间部分的1353NATY呈Ⅰ型β转角。C端的多功能停靠位点上的Tyr1349和Tyr1356是受体激酶活性的关键[2]。
目前已报道了大量的c-Met小分子抑制剂,部分抑制剂已进入临床研究阶段(见表1),本文根据其结合模式,将这些c-Met小分子抑制剂分为4类(Ⅰa型、Ⅰb型、Ⅱ型和其他类型)。
1 Ⅰa型c-Met选择性抑制剂
Ⅰa型c-Met抑制剂以U形构象结合在ATP结合位点,沿着铰链区向溶剂可及区延伸,可与c-Met激酶铰链区Pro1158、Met1160和Aspl222等氨基酸残基形成氢键,并与活化环上的Tyr1230形成π-π堆积作用,为ATP竞争性抑制剂。Ⅰa型c-Met抑制剂的细胞选择性大多相对较好,仅有少数会出现脱靶现象。

图1 HGF/c-MET信号转导通路Figure 1 HGF/c-Met signalling pathway
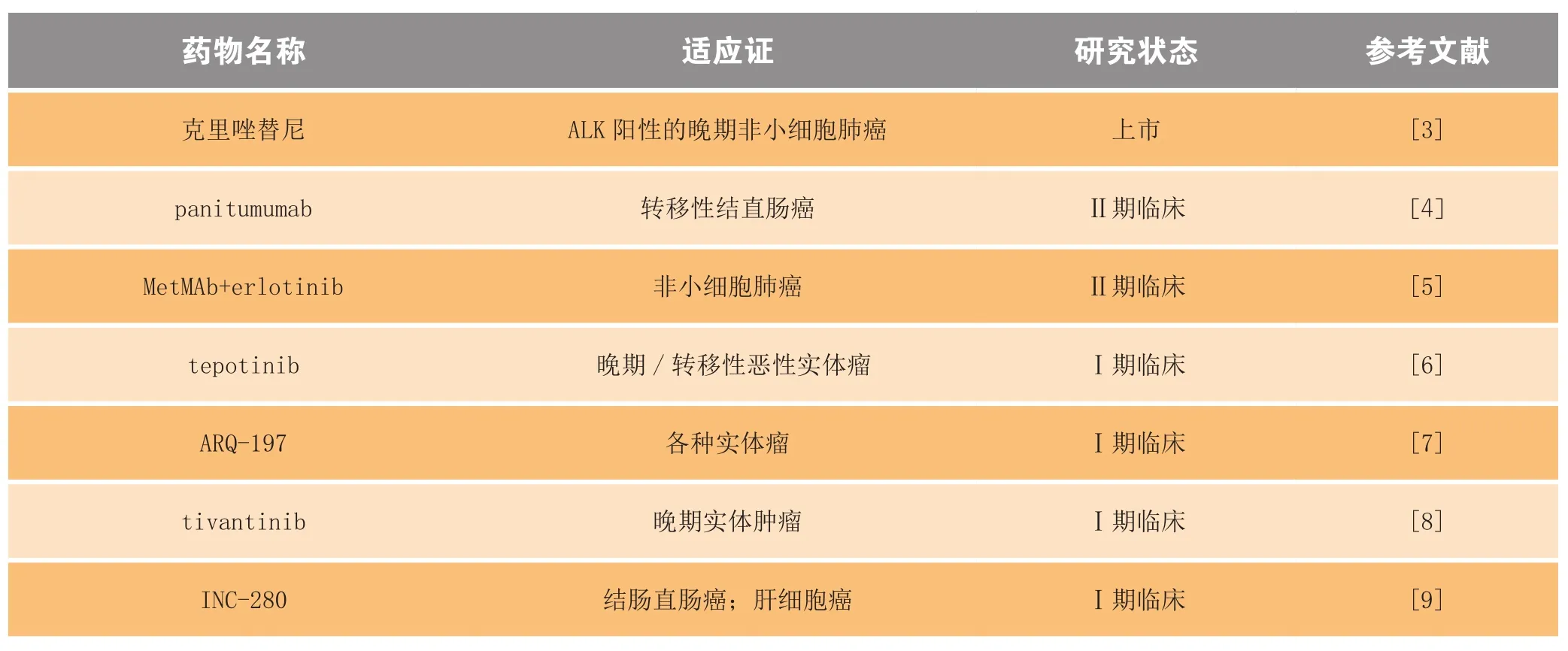
表1 部分进入临床研究阶段的小分子c-Met激酶抑制剂Table1 Some c-Met inhibitors currently undergoing clinical trials
1.1 吲哚-2-酮类化合物
3-取代吲哚-2-酮类化合物的代表性药物为舒尼替尼,其适应证为胃肠道间质瘤(gastrointestinal stromal tumor,GIST)和肾细胞癌(renal cell carcinoma,RCC)[10]。此类化合物对激酶的选择性主要依靠该母核结构上的取代基团。Sugen公司(现属于辉瑞公司)的研究人员通过对4-取代[11]和5-取代[12]的吲哚-2-酮类化合物的研究发现了化合物SU-11274(1),其对c-Met的IC50为10 nmol·L-1[13],针对GTL16细胞株的IC50为0.288 μmol·L-1。对SU11274的结构修饰又得到了PHA-665752(2),其细胞活性和选择性都得到了显著提高,其对c-Met的IC50为9 nmol·L-1,针对GTL16细胞株的IC50为9 nmol·L-1[14]。PHA-665752(2)的活性和选择性均高于SU11271(3)(IC50=40 nmol·L-1)、SU11205(4)(IC50=170 nmol·L-1)[13]。

1.2 2-氨基-5-芳基-3-苄氧基吡啶类化合物
辉瑞公司的研究人员通过对PHA-665752的结构改造,设计得到比吲哚-2-酮类化合物与非磷酸化状态下c-Met激酶域结合能力更强的2-氨基-5-芳基-3-苄氧基吡啶类化合物。研究人员以单芳香环的2-氨基吡啶替代了化合物PHA-665752中的吲哚-2酮与铰链区结合,并使柔性较好的3-苄氧基以更直接的角度进入化合物PHA-665752中2,6-二氯苯基所占据的疏水口袋,经优化得到c-Met、ALK、ROS1和RON选择性抑制剂克里唑替尼(PF-02341066,5)[14]。该药对ALK阳性的晚期非小细胞肺癌(non-small-cell lung cancer,NSCLC)患者疗效显著且安全性良好。体外研究显示,克里唑替尼能有效抑制c-Met磷酸化以及依赖c-Met进行的细胞增殖、转移和侵袭(IC50为5~20 nmol·L-1)。其晶体结合构象与PHA-665752的晶体结合构象相似,且其结构中的2,6-二氯-3-氟苯基占据了与PHA-665752的2,6-二氯苯基相同的空间位置并与A-loop的Tyr1230形成关键性的π-π堆积作用(见图2)[14]。
当细胞株与多种ALK变异体[如Karpas 299(NPM-ALK)、NCI-H3122(EML4-ALK变异体1)]或突变型[凯利神经母细胞瘤(突变型)]融合时,克里唑替尼还能通过抑制NSCLC中的ALK激酶与ATP的结合以及结合后的自身磷酸化而抑制激酶的激活,从而达到降低激酶活性和抗肿瘤作用,其对上述的ALK变异体或突变型的IC50为74~566 nmol·L-1[14]。图3为克里唑替尼与ALK激酶域复合物的晶体结构。
克里唑替尼的硫醚类似物(6)对c-Met的抑制活性有一定程度的下降(酶抑制活性:IC50=7.7 nmol·L-1;对SNU-5细胞株的抗增殖活性:IC50=190 nmol·L-1)[15]。化合物7是有高度选择性,耐受性良好,且口服有效的c-Met和ALK双重抑制剂,IC50分别为22 和39 nmol·L-1;化合物8(OSI-296)则是选择性的c-Met(IC50= nmol·L-1)和RON双重抑制剂,IC50分别为40和200 nmol·L-1,口服有效,耐受性良好,且具有良好的体内抗肿瘤活性[16]。
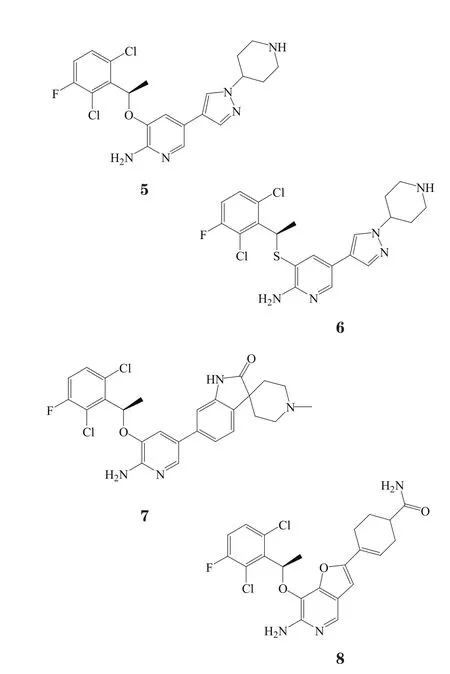

图2 PHA-665752和克里唑替尼与c-Met激酶复合物的晶体结构Figure 2 Co-crystal structures of PHA-665752/c-Met and crizotinib/c-Met
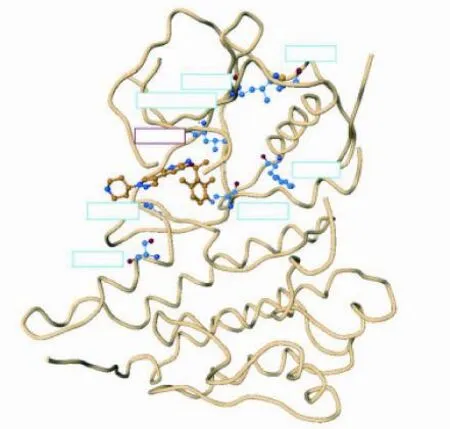
图3 克里唑替尼与ALK激酶域复合物的晶体结构Figure 3 Co-crystal structure of crizotinib /ALK kinase domain
1.3 其他类型化合物
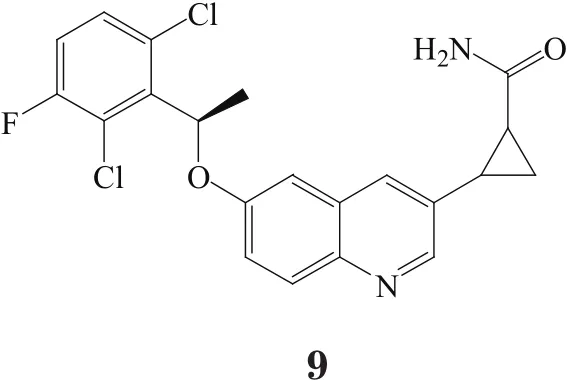
除了上述两类化合物外,还有几种新的结构类型保留了与Tyr1230形成关键性的π-π堆积作用的R-(2,6-二氯-3-氟苯基)甲基的Ⅰa型c-Met抑制剂。化合物9与化合物6的结合模式很相似,其对c-Met的IC50为9.3 nmol·L-1[17]。化合物10(X-376)对ALK的抑制活性(H3122细胞株中IC50为77 nmol·L-1)强于对c-Met(MKN-45细胞株中IC50为150 nmol·L-1)。化合物11利用吡啶酮作为铰链区结合基团,其疏水性比化合物10的铰链区结合基团低,所以它对c-Met的IC50为12 nmol·L-1,而对EBC-1细胞株的IC50为2 200 nmol·L-1[16]。
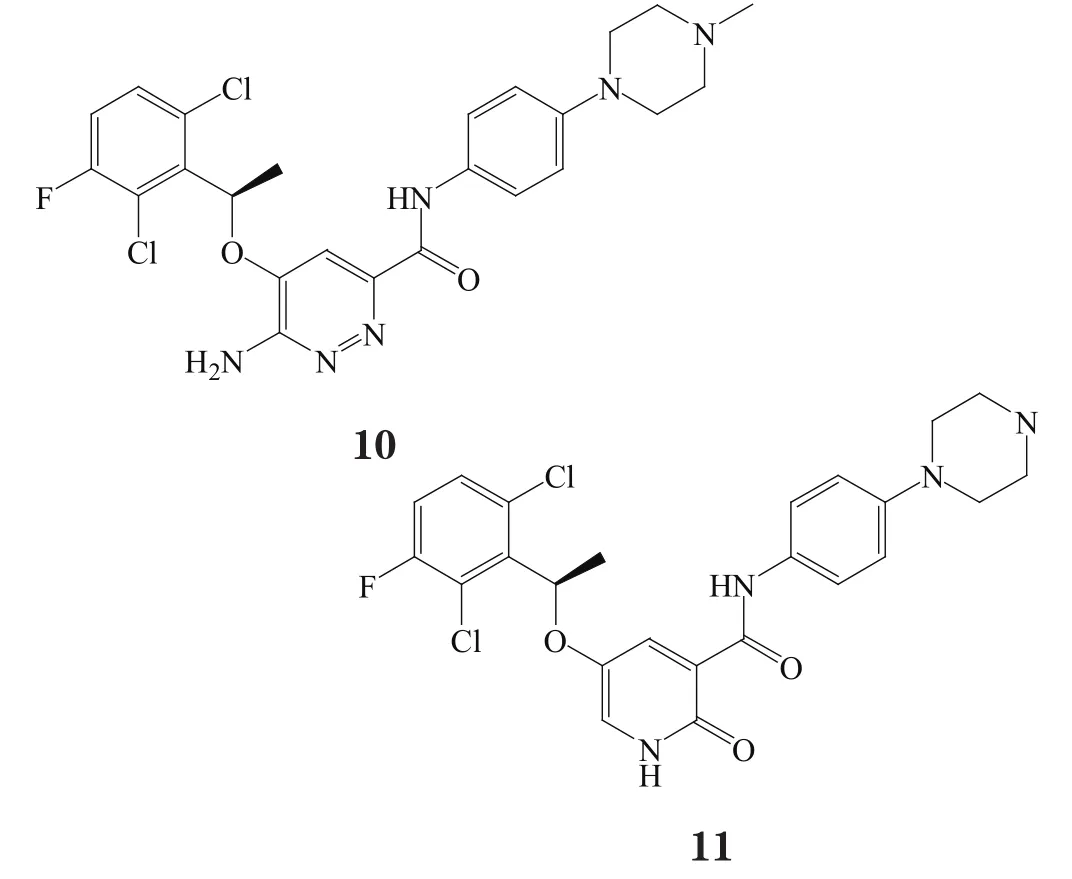
2 Ⅰb型c-Met选择性抑制剂
c-Met酪氨酸激酶的自抑制构象非常独特,在A-loop起始端有一个β折叠,且A-loop可直接与抑制剂产生相互作用,故其嘌呤结合位点比一般激酶更封闭。利用这种独特的结合位点设计的抑制剂即为Ⅰb型c-Met选择性抑制剂,其也是以U形构象结合于活性口袋,但与Ⅰa型抑制剂的延伸方向并不一致,前者主要沿着Asp1222、Tyr1230和Arg1208向溶剂可及区延伸,与Met1160、Asp1222和Arg1208形成氢键作用,并与Tyr1230也形成π-π堆积作用。独特结合特点使该类抑制剂具有高度的c-Met选择性。
Pharmacia公司通过高通量筛选的方法得到了高度选择性的c-Met抑制剂化合物12[18]。对其进行结构修饰所得到化合物13对c-Met激酶的Ki为1.3 nmol·L-1,也具有高度的c-Met选择性[19]。该化合物与非磷酸化状态下c-Met的复合物的晶体结构显示其酚羟基氧与铰链区Met1160的NH形成了氢键作用(见图4)[18],两者之间还通过结晶水分子形成了间接的氢键网络,化合物的亚甲基连接子还使得其酰肼-吲哚大共平面结构与A-loop的Tyr1230形成π-π堆积作用。
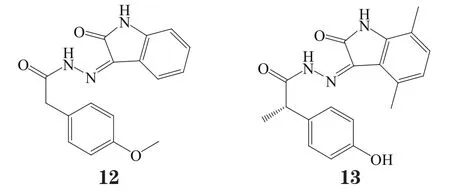

图4 化合物13与非磷酸化c-Met激酶域复合物的晶体结构Figure 4 Co-crystal structure of compound 13/c-Met kinase domain
Vojkovsky等[20]对化合物13进行结构改造,得到了以化合物14为代表的四元芳香环母核类化合物,其生物活性结果证明了骨架迁越的合理性。但由于这种四环结构的化合物的类药性较低,故被简化为以化合物15为代表的三氮唑三嗪双环母核。此类化合物的配体效率(Ligand efficiency, LE)比四环类抑制剂有所提高,但却不能与Arg1208的羰基形成氢键作用[21]。其双芳香环母核结构的缺电子性决定了其与Tyr1230之间的π-π堆积作用的强弱,这是该类化合物活性和选择性的一个决定性因素。因此,以三氮唑吡嗪(化合物16),三氮唑哒嗪(化合物17)为母核的化合物通常活性相对较低[21],对其结构优化改造得到化合物18(PF-04217903)[18]和化合物19(PF-04254644)[22]。这2个化合物体内外活性均较好,对c-Met激酶的Ki分别为5 和10.3 nmol·L-1,抑制肿瘤生长作用显著,口服药动学性质(PK)很好[18,23]。然而,化合物19对磷酸二酯酶有强抑制作用,存在心脏安全性问题,未能进入临床研究[22,24],而化合物18进入了临床研究阶段[23]。从化合物18与非磷酸化状态下的c-Met激酶域的复合物的晶体结构可看出,其与化合物13的晶体结合模式非常相似(见图5)[18]。
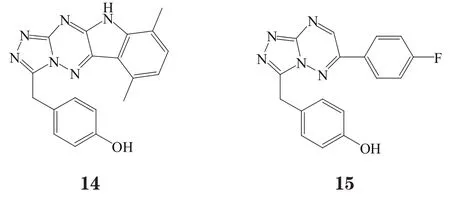

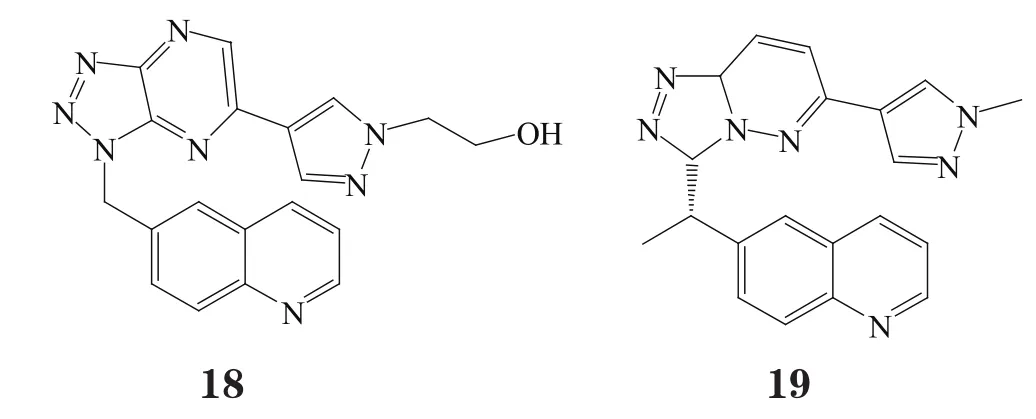

图5 PF-04217903与非磷酸化c-Met激酶域复合物的晶体结构Figure 5 Co-crystal structure of PF-04217903/c-Met kinase domain
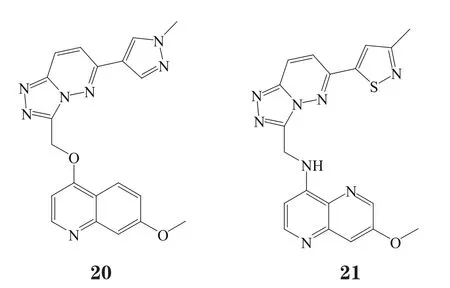
Albrecht等[25]以Sugen公司的专利化合物和Amgen公司的喹啉类c-Met抑制剂的结构为基础,通过结构片段重组的方法,得到化合物20(IC50=9 nmol·L-1),与上述Ⅰb型c-Me选择性抑制剂结构上的区别主要是喹啉的4位与连接子连接,并在连接子上增加了一个氧原子。对化合物20进行结构改造,用[1,5]萘啶替换喹啉,NH替换连接子上的氧,产生分子内氢键保持其优势构象,得到化合物21(IC50=5 nmol·L-1)[26]。从晶体复合物可看出,它们的结合模式与化合物13相同,可与A-loop上Tyr1230形成π-π堆积作用,故有较高的活性。
3 Ⅱ型c-Met非选择性抑制剂
在未活化的c-Met蛋白的自抑制构象的A-loop起始端有一个翻转,能够阻止抑制剂进入疏水性后口袋。Ⅰa型和Ⅰb型抑制剂能通过U形结合构象保持并稳固这种自抑制构象,从而表现出很高的结合力和选择性。Ⅱ型c-Met抑制剂通过“gatekeeper”氨基酸残基进入疏水性后口袋而发生作用,通常是多靶点的非c-Met选择性抑制剂。抑制剂进入后口袋必须使A-loop让出空间,这就要求Ⅱ型c-Met抑制剂有较高的相对分子质量和较强的疏水性,4-苯氧基喹啉是在VEGFRs、PDGFRs等多种激酶抑制剂中广泛被采用的母核结构。一些基于这种母核结构开发的c-Met抑制剂已于2003年申请专利[27]。化合物22对鳞状上皮细胞癌细胞(A431)中由人重组HGF诱导的c-Met磷酸化的抑制活性很高(IC50=0.008 7 μmol·L-1)。与PDGFR和VEGFR2的Ⅱ型抑制剂不同的是,该化合物中酰基硫脲连接子非常重要,它将最后的苯基延伸至疏水后口袋从而产生有效的c-Met抑制作用。
与VEGFR的Ⅱ型抑制剂相比,c-Met的Ⅱ型抑制剂一般具有更高的相对分子质量,配体效率较低,且是广谱的激酶抑制剂。例如,以环丙基-1,1-双甲酰胺作为连接子得到的卡博替尼(cabozatinib,23)便是一个多靶点激酶抑制剂,其对VEGFR2、c-Met、RET、KIT、FLT1/2/3、TIE-2及c-Met的 IC50分别为0.03、1.3、4、4.6、12/11.3/6、14.3和 7 nmol·L-1[28],对RON和PDGFR的活性稍弱,IC50分别为124和234 nmol·L-1。此外,在细胞活性测试中,卡博替尼也可有效抑制VEGFR2和c-Met的磷酸化,并对KIT、FLT3和AXL都有抑制作用,IC50分别为1.9、7.8、5.0、7.5和42 nmol·L-1,其在体内肿瘤模型中也能有效抑制c-Met和VEGFR2的磷酸化,并在临床前研究中表现出很好的抗肿瘤、抗转移以及抗血管生成活性[27]。总之,卡博替尼对c-Met和VEGFR信号转导通路调控异常的癌症具有非常好的疗效,能有效抑制肿瘤血管形成和肿瘤转移。该药于2012年11月29日被美国FDA批准用于治疗转移性甲状腺髓样癌[28]。

化合物24也是多靶点抑制剂,其对VEGFR2、c-Met、RON、KIT、FLT1/3/4、PDGFRα/β 和 TIE-2的IC50分别为0.86、0.4、3、6.7、6.8/3.6/2.8、3.6/9.6和1.1 nmol·L-1[29]。其与c-Met复合物的晶体结构显示4-氟苯胺基深入c-Met激酶后口袋,将Phe1223推出活性位点(见图6)。化合物25的铰链区结合基团是2-氨基吡啶,对c-Met和VEGFR2的IC50分别为14和16 nmol·L-1。
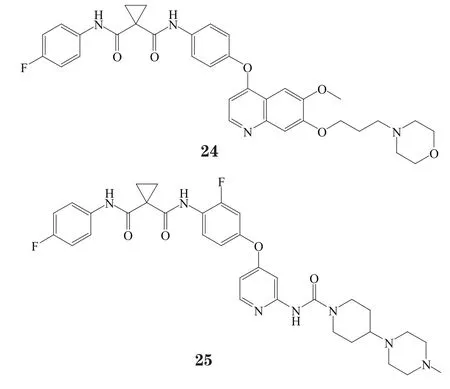

图6 化合物24与c-Met的相互作用表面Figure 6 c-Met protein surface that forms the binding pocket for compound 24
与Ⅰ型Met抑制剂相比,Ⅱ型Met抑制剂的配体亲脂性效率(lipophilic ligand efficiency, LLE)值更低,但脱靶更严重,尤其对蛋白激酶家族靶标。某些Ⅱ型抑制剂由于对VEGFR的活性比对c-Met更强,使其在未达到c-Met抑制所需剂量时就因VEGFR相关的副作用退出临床研究。
4 其他类型的c-Met抑制剂
化合物MK-2461(26)是一种新型的ATP竞争性多靶点抑制剂,作用于活化的c-Met,IC50为0.4~2.5 nmol·L-1,其对RON和FLT1作用效果稍弱,IC50分别为7和10 nmol·L-1;对c-Met的选择性是对FGFR1、FGFR2、FGFR3、PDGFRβ、KDR、FLT3、FLT4、TrkA和TrkB的8~30倍[30]。化合物26的一个类似物与活化的c-Met激酶晶体复合物结构显示该化合物完全占据了从嘌呤口袋到溶剂可及区的所有区域(见图7)。非磷酸化状态下c-Met独特的A-loop自抑制构象不适合与化合物26这样的共轭、共平面的母核结构结合,故化合物26对非活化状态下c-Met的抑制活性(Kd=27.2 nmol·L-1)明显比活化状态(Kd=4.4 nmol·L-1)的低[31]。


图7 化合物26的类似化合物与非磷酸化c-Met复合物的晶体结构Figure 7 Co-crystal structure of analogs of compound 26/c-Met kinase domain
在对转移性癌细胞的表型细胞活力检测时发现了化合物27,随后又证明c-Met是该化合物的作用靶标。该化合物是一个非ATP竞争性抑制剂,其对c-Met的Ki为355 nmol·L-1[32]。

5 结语
自1984年首次发现c-Met激酶,有关HGF/c-Met信号转导通路的生物功能及其与疾病的病理关系的研究和报道以指数级出现,尤其在最近10年内。HGF/c-Met信号转导通路活化在人类恶性肿瘤中的普遍存在促使靶向HGF/c-Met信号转导通路的药物发现项目快速发展。超过20个不同的治疗药物,包括HGF单克隆抗体、c-Met单克隆抗体以及c-Met激酶小分子抑制剂,已进入临床研究阶段。临床研究结果将最终确证HGF/c-Met在癌症中的病理作用并为肿瘤治疗提供全新的药物。
由于c-Met在细胞存活、生长、血管生成和转移过程中的作用,c-Met仍然是极具希望的抗肿瘤药物研发靶点,但将c-Met抑制剂开发成为癌症治疗药物仍然面临许多挑战,包括如何靶向识别并抑制c-Met而产生治疗效果、开发可用于准确诊断病情和分析药效的生物标记物并将之应用于临床,以及确定正确的联合治疗方法。在目前大量靶向HGF/c-Met的抑制剂的基础上,针对不同病情特点的临床研究将阐明HGF/c-Met信号转导通路在癌症进程中的作用,推动临床前研究向有效治疗策略的转化,加速c-Met抑制剂类药物的开发进程。
[1]Furge K A , Zhang Y W, Vande Woude G F, et al.c-Met receptor tyrosine kinase: enhanced signaling throughN adapter proteins [J].Oncogene, 2000, 19 ( 49 ): 5582- 558O9.
[2]Schiering N, Knapp S,O Marina M.CryNstal strNucture of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microNbial alOkaloid K2 252a[J].Proc Natl Acad Sci USA , 2003,10N0 ( 22 ): 1N2654-12659.
[3]Cui J J,Tran-DubOe M, Shen H, et al.Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-Met) kinase and anaplastic lymphoma kinase (ALK) [J].J Med Chem, 2011, 54 (18): 6342-6363.
[4]Eng C, Nowara E, Swieboda-Sadlej A, et al.Primary analysis and biomarker evaluation: randomized phase IB/II study of rilotumumab (AMG 102) or ganitumab (AMG 479) with panitumumab versus panitumumab alone in patients (pts) with metastatic colorectal cancer (mCRC)[J].Ann Oncol,2011, 22(Suppl 5): Abst O-0022
[5]Yu W, Pandita A, Penuel E, et al.Exploratory biomarker analyses from OAM4558g:a placebo-controlled phase II study of erlotinib with or without MetMAb in patients with advanced non-small cell lung cancer (NSCLC):47th Annu Meet Am Soc Clin Oncol (ASCO) ,Chicago,June 3-7, 2011[C].[S.l.]:[s.n.],c2011:Abst 7529.
[6]Falchook G,Hong D,Amin H,et al.First-in-human phase I trial assessing the highly selective c-Met inhibitor MSC2156119J (EMD 1214063) in patients with advanced solid tumors:39th Eur Soc Med Oncol (ESMO) Congr, Madrid ,September 26-30,2014[C].[S.l.]:[s.n.],c2014:Abst 450PD.
[7]Yap T A, Harris D, Barriuso J, et al.Phase I trial to determine the dose range for the c-Met inhibitor ARQ 197 that inhibits c-Met and FAK phosphorylation, when administered by an oral twice-a-day schedule[J].J Clin Oncol 2008, 26( Suppl15): Abst 3584.
[8]Sandhu S, Yap T, Tunariu N, et al.Pharmacokinetic and pharmacodynamic phase I trial of ARQ197 incorporating dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) studies investigating the antiangiogenic activity of selective c-Met inhibition:NCRI Cancer Conf, Birmingham,October 4-7,2009, [C].[S.l.]:[s.n.],c2009:AbstC141.
[9]Donehower R C, Scardina A, Hill M, et al.A phase I dose-escalation study of INCB028060, an inhibitor of c-MET receptor tyrosine kinase, in patients with advanced solid tumors:47th Annu Meet Am Soc Clin Oncol (ASCO),Chicago,June 3-7,2011 [C].[S.l.]:[s.n.],c2009:Abst 3091.
[10]Sun L, Liang C, Shirazian S, et al.Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenec-Methyl]-2,4- dic-Methyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase [J].J Med Chem, 2003, 46 (7): 1116-1119.
[11]Cui J, Zhang R, Shen H, et al.Preparation of 4-Aryl substituted indolinones as protein kinase signal transduction modulators for inhibiting abnormal cell proliferation: WO, 0255517[P].2002-07-18.
[12]Cui J, Ramphal Y, Liang C, et al.5-Aralkylsulfonyl-3-(pyrrol-2-ylc-Methylidene)-2-indolinone derivatives as kinase inhibitors:WO, 2002096361 A2[P].2002-12-05.
[13]Wang X, Le P, Liang C, et al.Potent and selective inhibitors of the c-Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion [J].Mol Cancer Ther, 2003, 2 (11): 1085-1092.
[14]Christensen J G, Schreck R, Burrows J, et al.A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo [J].Cancer Res, 2003, 63 (21): 7345-7355.
[15]Zhang D, Zhang X, Ai J, et al.Synthesis and biological evaluation of 2-amino-5-aryl-3-benzylthiopyridine scaffold based potent c-Met inhibitors [J].Bioorg Med Chem, 2013, 21 (21): 6804-6820.
[16]Steinig A G, Li A H, Wang J, et al.Novel 6-aminofuro[3, 2-c]pyridines as potent, orally effcacious inhibitors of c-Met and RON kinases [J].Bioorg Med Chem Lett, 2013, 23 (15): 4381-4387.
[17]Nishii H, Chiba-T, Morikami K, et al.Discovery of 6-benzyloxyquinolines as c-Met selective kinase inhibitors [J].Bioorg Med Chem Lett, 2010, 20 (4): 1405-1409.
[18]Cui J J, McTigue, M, Nambu M, et al.Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-Met) protein kinase inhibitors and identification of the clinical candidate2-(4-(1-(quinolin-6-ylc-Methyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer [J].J Med Chem, 2012, 55 (18): 8091-8109.
[19]Koenig M, Cui J, Wei C C, et al.Indolinone hydrazides as c-Met inhibitors:WO, 2005005378[P].2005-01-20.
[20]Vojkovsky T, Koenig M, Zhang F J, et al.Tetracyclic compounds as c-Met inhibitors:WO, 2005004808[P].2005-01-20.
[21]Cui J, Botrous I.Arylc-methyl triazolo and imidazopyrazines as c-Met inhibitors:WO, 2005004607[P].2005-01-20.
[22]Hu W, Hirakawa B, Jessen B, et al.A tyrosine kinase inhibitor-induced myocardial degeneration in rats through off-target phosphodiesterase inhibition [J].J Appl Toxicol, 2012, 32 (12): 1008-1020.
[23]Cui J J, Shen H, Tran-Dube M, et al.Lessons from 6-(1-(6-(1-c-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)ethyl) quinoline(PF-04254644), an inhibitor of receptor tyrosine kinase c-Met with high protein kinase selectivity but broad phosphodiesterase family inhibition leading to myocardial degeneration in rats [J].J Med Chem, 2013, 56 (17): 6651-6665.
[24]Aguirre S A, Heyen J R, Collette W 3rd, et al.Cardiovascular effects in rats following exposure to a receptor tyrosine kinase inhibitor [J].Toxicol Pathol, 2010, 38 (3): 416-428.
[25]Albrecht B K, Harmange J C, Bauer D, et al.Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase [J].J Med Chem, 2008, 51 (10): 2879-2882.
[26]Boezio A A, Berry L, Albrecht B K, et al.Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors [J].Bioorg Med Chem Lett, 2009, 19 (22): 6307-6312.
[27]Yakes F M, Chen J, Tan J, et al.Cabozantinib (XL184), a novel c-Met and VEGFR2 inhibitor, simultaneously suppresses c-Metastasis, angiogenesis, and tumor growth [J].Mol Cancer Ther, 2011, 10 (12):2298-2308.
[28]Fujiwara Y, Miwa A, Nakamura K, et al.Quinoline derivative and quinazoline derivative inhibiting self-phosphorylation of hepatocytus proliferator receptor, and medicinal composition containing the same:WO, 2003000660[P].2003-01-03.
[29]Qian F, Engst S, Yamaguchi K, et al.Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases [J].Cancer Res, 2009, 69 (20): 8009-8016.
[30]Pan B S, Chan G K, Chenard M, et al.MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor [J].Cancer Res, 2010, 70 (4): 1524-1533.
[31]Sadiq A A, Salgia R.c-Met as a possible target for non-small-cell lung cancer [J].J Clin Oncol, 2013, 31 (8): 1089-1096.
[32]Munshi N, Jeay S, Li Y, et al.ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity [J].Mol Cancer Ther, 2010, 9 (6): 1544-1553.
Advances in Research on c-Met Kinase Inhibitors
HU Yaning1, HU Shihe1, YUAN Haoliang1, ZHUANG Jin1, CHEN Yadong2, LU Tao1
(1.Division of Organic Chemistry, China Pharmaceutical University, Nanjing 211198, China; 2.Division of Inorganic Chemistry, China Pharmaceutical University, Nanjing 211198, China)
c-Met kinase is the receptor for hepatocyte growth factor(HGF).HGF/c-Met is frequently activated during the development, growth and metastasis of tumor.Therefore, c-Met becomes an important target in anti-cancer therapy.The advances in research on c-Met inhibitors recent years have been reviewed in this paper.
c-Met kinase; HGF/c-Met signalling pathway; c-Met inhibitor; tumor
R914.5
A
1001-5094(2015)03-0170-09
接受日期:2015-02-06
项目资助:国家自然科学基金( No.81473078) ;
*通讯作者:陆涛,教授;
研究方向:新药分子设计与合成研究,计算机辅助药物设计;
Tel:025-83271086;E-mail:lutao@cpu.edu.cn
