顶空固相微萃取-气相色谱法测定N,N-二甲基羟胺
2015-01-17吴继宗邓惟勤陶苗苗谈树苹
王 玮,吴继宗,邓惟勤,陶苗苗,谈树苹
中国原子能科学研究院 放射化学研究所,北京 102413
顶空固相微萃取-气相色谱法
测定N,N-二甲基羟胺
王 玮,吴继宗,邓惟勤,陶苗苗,谈树苹
中国原子能科学研究院 放射化学研究所,北京 102413
建立了顶空固相微萃取-气相色谱测定N,N-二甲基羟胺(DMHAN)的分析方法。采用65 μm聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)进行萃取,考察了萃取时间、萃取温度、溶液pH值、顶空体积及解吸时间等因素的影响,确定了顶空固相微萃取-气相色谱分析DMHAN的最佳实验条件。DMHAN浓度在1.65~49.4 mmol/L范围内有良好的线性关系(r=0.997 3),当浓度为8.24 mmol/L时相对标准偏差为2.1%(n=5),检出限为4.67×10-2mmol/L,重加回收率为96.1%~103%。该方法操作简单、稳定,适合后处理料液中DMHAN的测定。
N,N-二甲基羟胺;顶空固相微萃取;气相色谱法
在现代Purex流程的发展中, 随着核燃料燃耗的加深及对废物最小化的要求, 新型无盐还原剂的研究成为改善流程的一个重要方向[1]。N,N-二甲基羟胺(DMHAN)比传统的无盐还原剂羟胺还原速率快,反应完全[2], 近年来其研究和应用备受重视。
目前,工艺中主要采用氧化还原-间接分光光度法[3]分析DMHAN,然而由于后处理料液中同时存在甲基肼、甲酸、甲醛等还原性组分[4]会干扰DMHAN的测定。李高亮等[5]采用气相色谱法分析DMHAN,该方法采用液体直接进样。由于后处理料液多为硝酸溶液,为了避免损坏色谱柱,须采用氢氧化钠溶液调节样品pH至略大于7;对于含钚样品测定时,首先要用N,N,N′,N′-四辛基-3-氧戊二酰胺(TODGA)萃取出钚。萃取钚的操作步骤繁琐,消耗较多有机溶剂的同时会产生更多放射性废液。因此,需要建立一个更加简单有效测定DMHAN的方法。
固相微萃取(SPME)技术是在固相萃取基础上发展起来的一种新的萃取分离技术,具有操作简单,无需使用有机溶剂,适用于直接采集挥发性和半挥发性的物质,萃取样品后可直接色谱进样等优点[6]。由于DMHAN属于半挥发性物质,可把固相微萃取技术用于DMHAN的分析,将固相微萃取头置于样品上部空间(顶空方式)进行萃取,可以排除样品基体对测定的干扰和仪器沾污。本工作拟建立顶空固相微萃取-气相色谱测定DMHAN的方法。
1 实验部分
1.1 试剂和仪器
N,N-二甲基羟胺盐酸盐,纯度99%,美国西格玛公司;5.4 mol/LN,N-二甲基羟胺的水溶液,纯度大于99%,中国原子能科学研究院放射化学研究所;无水碳酸钠、硝酸均为分析纯,北京化学试剂二厂。
GC-2010PLUS型气相色谱仪,日本岛津公司;GH-300型超纯氢气发生器,北京中兴汇利科技发展有限公司;WYB-2型静音无油空气泵,北京中亚气体仪器研究所;凹槽式加热器(23 mm×75 mm,20 mL顶空瓶专用),上海赛鹭鑫分析技术有限公司;SPME装置(装置示于图1,包括一个萃取手柄,PDMS/DVB萃取头),Supelco公司;20 mL顶空进样瓶(23 mm×75 mm),铝盖+硅胶垫(配20 mL顶空瓶),压盖器、启盖器,Thermo Fisher公司;Seven Multi 多功能pH计、ML204/02型电子天平(精度0.1 mg),METTLER TOLEDO公司。

1——推杆(Plunger),2——手柄筒((Barrel),3——Z型槽(“Z” slot),4——支撑推杆旋钮(Plunger retaining screw),5——透视窗口(Viewing window)6——可调节深度计(Adjustable needle guide)7——SPME萃取头(SPME fiber)8——穿孔针(Septum-piercing needle)9——纤维连接管(Fiber-attachment needle)10——涂层石英纤维(Coated SPME fused silica fiber)图1 固相微萃取装置Fig.1 SPME device
1.2 实验方法

图2 顶空固相微萃取操作步骤Fig.2 Operating steps of HS-SPME
将待测样品置于20 mL顶空瓶中,根据需要加入试剂调节溶液pH,或加入去离子水稀释待测样品后,将顶空瓶密封,插入SPME萃取头于顶空体积中,流程示于图2。在一定温度下, SPME萃取头的表面涂层对气相中各组分进行选择性吸附,DMHAN在萃取头表面富集,经过一定时间,DMHAN的含量将在液相、气相和固相(SPME萃取涂层)三相中达到平衡[7]。最后将SPME萃取头取出,插入气相色谱仪进行分析。
2 结果与讨论
2.1 色谱条件选择
2.1.1 进样口温度 采用固相微萃取进样时,既要保证萃取头上吸附的待测物在气相色谱仪的进样口能够快速、完全地解吸下来,又要避免因温度过高造成待测物分解,进样口温度(即待测物从萃取头上解吸下来的温度)是关键的色谱条件。考察了进样口温度分别为130、150、180、200、235 ℃时,DMHAN的热解吸情况。当进样口温度高于180 ℃时,色谱图上出现峰面积较大的杂峰;而当进样口温度降到130 ℃时,杂峰消失。因此本实验采用较低温度进行热解吸。这是由于DMHAN的沸点并不高,采用较低的进样口温度就可以保证萃取头上吸附的待测物热解吸完全。为了验证萃取头上是否有残留,不改变其它操作条件的前提下将进样口温度升至235 ℃,把实验后的SPME萃取头插入进样口热解吸进样分析,没有色谱峰出现,说明130 ℃可以保证热解吸完全。最终,进样口温度定为130 ℃,在此温度下热解吸既可以保证DMHAN峰形较好,也能够完全解吸无残留。
2.1.2 柱温 由于DMHAN极性较强,本工作选择强极性的FFAP毛细管柱对其及其它可挥发性组分进行分离。柱温设为文献[5]中的120 ℃时,DMHAN出峰较快,保留时间为1.49 min,但样品中共存的甲醇、乙醇与其保留时间相同,无法分析。实验中逐步将柱温从120 ℃降到90 ℃后可以实现DMHAN与甲醇、乙醇等完全分离。因此,选择柱温90 ℃分析DMHAN。
综上所述,分析DMHAN的色谱条件确定为:FFAP毛细管柱(内径0.25 mm,膜厚0.25 μm,柱长30 m);载气为氮气,线速率21 cm/s;分流进样,分流比15∶1;进样口温度130 ℃,柱温90 ℃;FID检测器温度150 ℃,氢气流量40 mL/min,空气流量400 mL/min。在选定的色谱条件下,分析DMHAN与甲醇、乙醇、甲酸、甲胺、甲醛、甲基肼的混合溶液,结果示于图3。由图3可知,样品中只有甲醇、乙醇、DMHAN出峰,保留时间分别为2.75、2.83、3.28 min, 甲醇、乙醇与DMHAN的色谱峰分离较好, 不会影响其定性定量分析。

图3 N,N-二甲基羟胺与甲醇、乙醇、甲酸、甲胺、甲醛、甲基肼混合溶液的色谱图Fig.3 Gas chromatogram of mixture of DMHAN, methanol, ethanol, methanoic acid, methylamine, formaldehyde, monomethylhydrazine
2.2 顶空固相微萃取条件选择
比较了65 μm聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)涂层和85 μm聚丙烯酸酯(PA)涂层对N,N-二甲基羟胺的萃取效果,实验结果表明:PDMS/DVB涂层对N,N-二甲基羟胺萃取效果好,而PA萃取涂层无法萃取N,N-二甲基羟胺。由于PDMS/DVB涂层对胺类化合物的吸附和解吸的效率较高[8],因此本实验选用65 μm PDMS/DVB萃取头。
2.2.1 萃取时间 取5 mL 12.0 mmol/L的N,N-二甲基羟胺水溶液加入到20 mL顶空瓶中,密封后放入预热温度为25 ℃(室温10 ℃)的加热器凹槽中,按照顶空固相微萃取操作步骤测定5次,考察不同萃取时间10、20、30、40、50 min对N,N-二甲基羟胺峰面积的影响。结果示于图4。由图4可知:在起始阶段,DMHAN峰面积随顶空固相微萃取时间的延长迅速增加;当萃取时间达到20 min后,DMHAN峰面积趋于不变。因此,本实验应选择萃取时间为20 min。
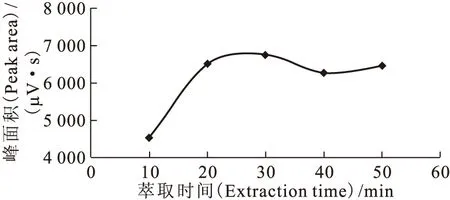
图4 萃取时间的影响Fig.4 Effect of extraction time
2.2.2 萃取温度 由2.2.1节可知:萃取温度为25 ℃时,萃取时间为20 min,DMHAN即可在SPME萃取涂层上达到吸附平衡。升高温度不仅可以促进DMHAN逸出,提高其在气相中的浓度,而且加快了传质,有利于缩短平衡时间;但温度过高可能会导致DMHAN稳定性和其在萃取涂层上分配系数的降低。取12.0 mmol/L的N,N-二甲基羟胺水溶液5 mL于20 mL顶空瓶中密封,分别在20、25、30、40、50、60 ℃下萃取,选用65 μm的PDMS/DVB萃取头顶空萃取20 min后热解吸进样,考察萃取温度对萃取效果的影响,结果示于图5。由图5可知:N,N-二甲基羟胺的峰面积在20~40 ℃随温度升高而增大,当温度高于40 ℃时峰面积趋于不变;当温度高于40 ℃时,N,N-二甲基羟胺色谱峰峰形随温度升高而变差,并出现峰面积较大的未知峰。故本实验选择最佳萃取温度为40 ℃。
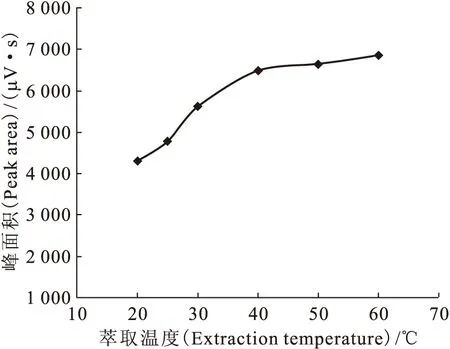
图5 萃取温度的影响Fig.5 Effect of extraction temperature
2.2.3 样品溶液的pH值 硝酸体系中的N,N-二甲基羟胺以硝酸盐的形式存在,加入过量饱和碳酸钠溶液与样品中的硝酸反应可以提高其挥发性。考察了不同pH值条件下65 μm PDMS/DVB涂层对N,N-二甲基羟胺的萃取效果,结果列入表1。由表1可知:随着溶液的pH值增大,色谱测定峰面积增大,说明有更多的N,N-二甲基羟胺挥发到顶空体系。过量饱和碳酸钠溶液可调节待测样品溶液pH至10.3~10.5,此时N,N-二甲基羟胺的峰面积最大。而且使用饱和碳酸钠溶液调节溶液pH值,操作简单方便,可维持每次测定所调节的pH值基本相同。因此,本实验选择加入过量饱和碳酸钠溶液调节样品溶液的pH值。
2.2.4 顶空体积 顶空瓶中样品与样品上方的气体体积的比值(相比)是影响顶空分析灵敏度的关键因素[9]。本实验选用20 mL顶空瓶,在以上已经确定的实验条件下,分别考察了样品溶液为2.5、5 mL和10 mL时12.0 mmol/L的N,N-二甲基羟胺萃取效果的变化(3份样品待测物实际浓度相同,只改变待测样品的体积),得到实验结果列入表2。由表2可知:随着顶空瓶中样品体积的增加即相比的增加,顶空体积中待测物浓度增大,N,N-二甲基羟胺的峰面积逐步提高。但是兼顾到后处理体系中的样品具有一定的放射性、基体组成复杂,把每次分析样品的体积定为5 mL既可以满足方法灵敏度的要求,又利于节约试剂和减少废液。因此,最终选定使用20 mL顶空瓶测定时,样品体积为5 mL。

表1 pH值的影响Table 1 Effect of pH

表2 顶空体积的影响Table 2 Effect of headspace volume
2.2.5 解吸时间 本实验在气相色谱仪进样口温度为130 ℃ 下热解吸进样, 若解吸时间过短有可能解吸不完全,残留的待测物会影响下一次的分析。考察了解吸时间为1、2、3、4 min的解吸情况,结果表明:N,N-二甲基羟胺3 min即可解吸完全。因此选择解吸时间为3 min。
2.3 标准工作曲线及检出限
准确吸取适量N,N-二甲基羟胺标准储备液,配制浓度分别为1.65、4.12、8.24、16.5、24.7、33.0、49.4 mmol/L 的N,N-二甲基羟胺系列标准溶液,在上述已优化的顶空固相微萃取条件下,按2.1.2节的色谱条件测定N,N-二甲基羟胺色谱峰面积,绘制其浓度c与色谱峰面积A的标准工作曲线示于图6。由图6可知:N,N-二甲基羟胺浓度在1.65~49.4 mmol/L线性良好,回归方程为y=3 297.9x+8 805.9,相关系数r=0.997 3。在2.1.2节色谱条件下测定N,N-二甲基羟胺,记录基线噪声,按3倍基线噪声相当的量计算得出N,N-二甲基羟胺的检出限为4.67×10-2mmol/L。
2.4 精密度与准确度
DMHAN还原反萃Pu时,1BP中钚质量浓度约为 5 g/L、ρ(U)≈2.5 g/L,按照1BP中N,N-二甲基羟胺的浓度标准,需要将样品稀释20倍后测定,即钚约为 0.25 g/L,铀约为 0.125 g/L,使用硝酸亚铈、硝酸铀酰模拟代替后处理料液中的钚和铀,在每份待测样品中添加铈0.25 g/L、铀0.125 g/L。配制8.24 mmol/LN,N-二甲基羟胺待测样品共5份,按已优化的条件进行顶空固相微萃取和色谱测定,记录N,N-二甲基羟胺色谱峰面积,应用回归方程计算样品中N,N-二甲基羟胺浓度,计算sr值,结果列入表3。

图6 DMHAN峰面积与浓度的标准曲线Fig.6 DMHAN correction curve of peak area to concentration

表3 方法的精密度Table 3 Results of precision test
另取3份浓度为8.24 mmol/L的N,N-二甲基羟胺待测样品溶液(维持待测样品体积为5 mL时的实际浓度),添加4.12 mmol/L(待测样品体积为5 mL时的实际添加浓度)的N,N-二甲基羟胺标准溶液,得到回收率的实验结果列入表4。由表3、4可知:当N,N-二甲基羟胺浓度为8.24 mmol/L时,重复测定5次的相对标准偏差为2.1%;N,N-二甲基羟胺的重加回收率在96.1%~103%。

表4 方法的回收率Table 4 Results of recovery test
在我国先进后处理流程中,1BP中N,N-二甲基羟胺浓度约为0.075~0.08 mol/L,使用此方法时,将待测样品稀释20倍,即取样0.25 mL,加入饱和碳酸钠溶液1 mL,再加入3.75 mL去离子水后测定。对于其它工艺样品也可以通过稀释或减少取样量,使分析结果落入此方法的线性范围内。
3 结 论
顶空固相微萃取气相色谱分析N,N-二甲基羟胺,样品用量少,操作简单、稳定。在已优化的顶空固相微萃取和气相色谱条件下测定N,N-二甲基羟胺,浓度在1.65~49.4 mmol/L 之间线性良好(r=0.997 3);当浓度为8.24 mmol/L时,重复测定5次的相对标准偏差为2.1%,检出限为4.67×10-2mmol/L ,重加回收率为96.1%~103%,能够满足后处理料液中N,N-二甲基羟胺分析的要求。
[1] 肖松涛,欧阳应根.间接分光光度法测定单甲基肼、二甲基羟胺[R].中国核科技报告,2008(1):68-72.
[2] 何辉,胡景炘,张先业,等.N,N-二甲基羟胺对Pu(Ⅳ)的还原反萃和相应的计算机模型[J].核化学与放射化学,2001,23(2):65-71.
[3] 李传博,刘金平,晏太红,等.Purex流程中二甲基羟胺与甲基肼的分析[J].核化学与放射化学,2011,33(5):268-273.
[4] 陈辉.N,N-二甲基羟胺、甲基肼辐解行为和机理研究[D].北京:中国原子能科学研究院,2010.
[5] 李高亮,何辉,陈辉,等.N,N-二甲基羟胺的气相色谱分析[J].化学试剂,2008,30(7):515-516;549.
[6] 傅若农.固相微萃取(SPME)的演变和现状[J].化学试剂,2008,30(1):13-22.
[7] 荆瑞俊,李英,李永芳,等.顶空固相微萃取-气相色谱联用法测定工业废水中的致癌芳香胺[J].光谱实验室,2012,29(2):1141-1144.
[8] Zander A, Bishop A G, Prenzler P D. A solid phase microextraction method to fingerprint dissolved organic carbon released from Eucalyptus camaldulensis (Dehnh.)(River Red Gum) leaves[J]. Anal Chim Acta, 2005, 530(2): 325-333.
[9] 王立,汪正范编著.色谱分析样品处理[M].北京:化学工业出版社,2006.
Determination ofN,N-Dimethylhydroxylamine by Headspace Solid-Phase Micro-Extraction and Gas Chromatography
WANG Wei, WU Ji-zong, DENG Wei-qin, TAO Miao-miao, TAN Shu-ping
China Institute of Atomic Energy, P. O. Box 275(88), Beijing 102413, China
A method for the determination ofN,N-dimethylhydroxylamine(DMHAN) has been established by coupling headspace solid-phase micro-extraction (HS-SPME) with gas chromatography(GC). The 65 μm PDMS/DVB coating was selected for micro-extractor. The extraction conditions, such as extraction time, extraction temperature, solution acidity, headspace volume and desorption time were discussed and optimized. The linear range of DMHAN was 1.65-49.4 mmol/L (r=0.997 3). The relative standard deviation was 2.1% (n=5). The detection limit is 4.67×10-2mmol/L. The recoveries are from 96.1% to 103%. This method is simple, stable and suitable for the determination of DMHAN in the process solution of spent fuel reprocessing.
N,N-dimethylhydroxylamine; headspace solid-phase micro-extraction; gas chromatography
2014-06-16;
2014-07-18
王 玮(1987—),河北沧州人,硕士研究生,分析化学专业
TL283
A
0253-9950(2015)06-0441-06
10.7538/hhx.2015.37.06.0441
