维A酸软膏中维A酸的含量测定及有关物质的检查
2015-01-05解放军总医院药品保障中心北京100853
蔡 乐,姚 兰,白 林(解放军总医院药品保障中心,北京 100853)
·实验研究·
维A酸软膏中维A酸的含量测定及有关物质的检查
蔡 乐,姚 兰,白 林(解放军总医院药品保障中心,北京 100853)
目的:建立维A酸软膏中维A酸的含量测定及有关物质检查的方法。方法:采用HPLC法测定维A酸的含量。色谱柱:Agilent Zorbax SB C18柱(250 mm×4.6 mm,5 μm),流动相:甲醇-2%冰醋酸(85 : 15),流速:1.0 mL·min-1,检测波长:354 nm,柱温:45 ℃。结果:维A酸与其他杂质分离良好,与异维A酸的分离度> 5。维A酸在0.934 4 ~ 18.687 2 μg·mL-1范围内线性关系良好(r = 1.000 0);维A酸的平均回收率为99.4%(RSD = 1.44%,n = 9)。结论:采用该方法测定维A酸软膏中维A酸的含量操作简单,结果准确,可作为该制剂的质量控制方法。
高效液相色谱法;维A酸软膏;含量测定;维A酸;异维A酸
维A酸软膏收载于《中国人民解放军医疗机构制剂规范》(2002年版)[1],由维A酸、凡士林和液体石蜡组成,其中维A酸是维生素A的活性代谢产物,具有促进表皮细胞更新、调节表皮细胞增殖和分化作用,该药常用于各种类型的扁平苔藓、毛囊角化病、痤疮及其他角化异常类皮肤病的治疗。在《中国人民解放军医疗机构制剂规范》(2002年版)中并未记载维A酸软膏的含量测定方法,维A酸不稳定见光易转化为其顺势异构体异维A酸,为提高维A酸软膏的质量标准,更好地控制该制剂的质量,提高临床用药的安全性和有效性,本研究建立了测定维A酸软膏中维A酸的含量及检查有关物质异维A酸的HPLC方法。
1 仪器与试药
Agilent 1200型高效液相色谱仪(包括1200系列在线脱气机、四元梯度泵、自动进样器、柱温箱、VWD检测器和色谱工作站,美国Agilent公司);AG-285型电子天平(瑞士Mettler公司);UV-2550型紫外分光光度仪(日本岛津公司)。
维A酸对照品(中国食品药品检定研究院,纯度99.4%,批号100224-200903);异维A酸对照品(中国食品药品检定研究院,纯度99.8%,批号100224-200903);维A酸软膏(本院自制,规格:0.025%、20 g/盒,批号20140120-1、20140120-2、20140121);甲醇为色谱纯;乙醇、冰醋酸为分析纯;水为重蒸馏水。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Zorbax SB C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇-2%冰醋酸(85 : 15);流速:1.0 mL·min-1;检测波长:354 nm;柱温:45 ℃;进样量:10 μL。
2.2 溶液的制备
2.2.1对照品溶液 精密称定维A酸对照品9.4 mg,置100 mL棕色量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为维A酸对照品储备液。精密称定异维A酸对照品12.3 mg,置100 mL棕色量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为异维A酸对照品储备液A。精密量取异维A酸对照品储备液A 1 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,作为异维A酸对照品储备液B。精密量取维A酸对照品储备液2.5 mL和异维A酸对照品储备液B 4 mL,置100 mL量瓶中,加流动相稀释至刻度,摇匀,即得对照品溶液(含维A酸2.335 9 μg·mL-1和异维A酸0.491 0 μg·mL-1)。
2.2.2供试品溶液 避光操作,取供试品约1.0 g(含维A酸约0.25 mg),精密称定,置具塞锥形瓶中,精密加入无水乙醇50 mL,称定重量,于60 ℃水浴中充分振摇15 min,再置于冰浴中冷却2 h。取出后用无水乙醇补足重量,迅速滤过,放至室温后,取续滤液5 mL置10 mL棕色量瓶中,加流动相稀释至刻度,摇匀,用0.22 μm微孔滤膜滤过,取续滤液,作为供试品溶液。
2.2.3阴性对照溶液 另按处方比例及制备工艺,取除维A酸以外的其余成分,配制不含维A酸的空白制剂,依供试品溶液制备方法制备阴性对照溶液。
2.3 系统适用性实验
分别取对照品溶液、供试品溶液、阴性对照溶液按“2.1”项下色谱条件,各进样10 μL,测定HPLC图谱,考察供试品中维A酸、异维A酸和其他组分色谱峰分离情况。在此色谱条件下,维A酸的保留时间为18.4 min,异维A酸的保留时间为16.3 min,维A酸和异维A酸色谱峰峰形对称尖锐,分离度大于5,基线平稳,与其他色谱峰分离良好,阴性对照无干扰,见图1。
2.4 线性关系考察
精密量取维A酸储备液适量,加流动相配制成含维A酸分别为0.934 4、2.335 9、4.671 8、9.343 6、14.015 4、18.687 2 μg·mL-1的标准溶液。分别精密吸取标准溶液各10 μL,按“2.1”项下色谱条件进行测定。以峰面积值为纵坐标(Y),浓度为横坐标(X)绘制标准曲线,计算得维A酸的回归方程为Y = 89.383 X + 1.874 2,r = 1.000 0。结果表明,维A酸在0.934 4 ~18.687 2 µg·mL-1范围内与峰面积呈良好线性关系。

图1 维A酸软膏的HPLC色谱图A – 阴性对照,B – 对照品,C – 供试品;1 – 异维A酸,2 – 维A酸Fig 1 HPLC chromatogram of tretinoin ointmentA – negative reference substance, B – reference substance, C – sample; 1 – isotretinoin, 2 – tretinoin
2.5 精密度实验
取“2.2.1”项下对照品溶液,按“2.1”项下色谱条件,连续进样6次测定,计算维A酸的峰面积RSD为0.06%(n = 6),表明仪器精密度良好。
2.6 重复性实验
取同一供试品(批号20140120-1)约1.0 g,共6份,精密称定,分别按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进行测定,计算含量RSD为1.73%(n = 6),表明方法重复性良好。
2.7 回收率实验
取“2.2.3”项下空白制剂9份,分成3组,每组3份,每份约1.0 g。分别精密加入一定量的维A酸对照品储备液,于50 ℃微温下与空白制剂混合,再按供试品溶液制备方法制成含量约为供试品标示量的80%、100%、120%的样品溶液。按“2.1”项下色谱条件进行测定,计算回收率,结果见表1。
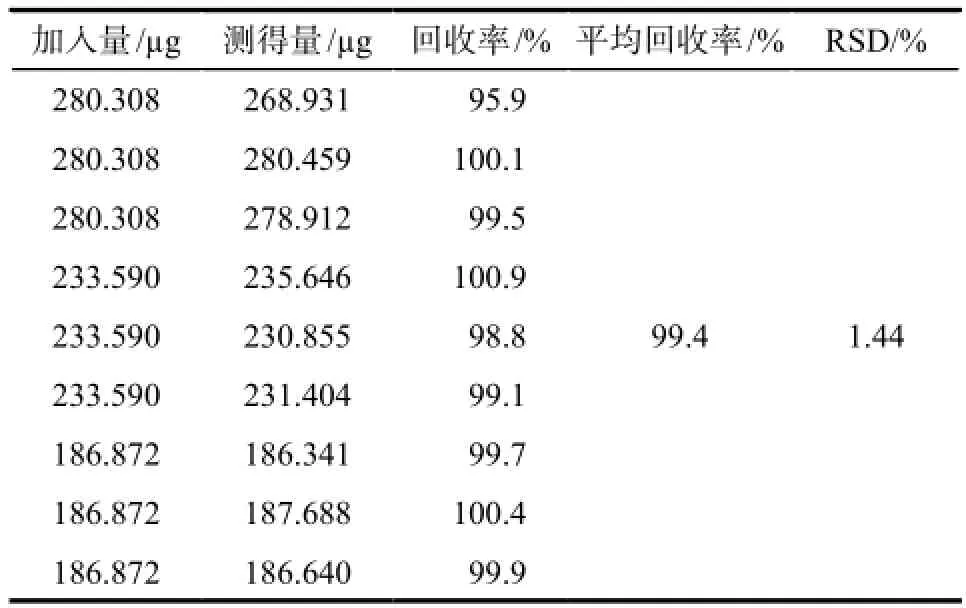
表1 维A酸软膏回收率实验测定结果.n= 9Tab 1 Results of recovery test about tretinoin ointment.n= 9
2.8 样品含量测定
取3批样品(批号20140120-1、20140120-2、20140121),每批3份,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进行测定,以外标法计算各个样品中维A酸的含量,结果见表2。

表2 维A酸软膏中维A酸的含量.n= 3Tab 2 Determination of contents of tretinoin in tretinoin ointment.n= 3
2.9 异维A酸限量检查
取3批样品(批号20140120-1、20140120-2、20140121),按“2.2.2”项下方法制备供试品溶液,依照“2.1”项下色谱条件,40 μL进样测定。供试品溶液色谱图中若有与异维A酸保留时间一致的色谱峰,其峰面积不得大于对照品溶液10 μL进样时异维A酸峰面积的5%。经测定,3批样品异维A酸的限量均符合规定。
3 讨论
3.1 维A酸含量测定方法的选择
维A酸的含量测定方法主要有UV法[2-3]和HPLC法[4-6]。经实验发现,维A酸软膏中凡士林等基质和异维A酸等杂质在维A酸最大吸收波长处均有吸收,对紫外分光光度法测定存在影响,采用HPLC法则可避免基质和杂质对维A酸测定的干扰,并可对制剂中的杂质进行限量检查。由于软膏基质黏度大,成分复杂,处理不当易造成色谱柱堵塞。因此,本实验在参考文献基础上,对提取条件等进行了优化。
3.2 色谱条件的选择
研究中曾参考文献[6]维A酸原料药含量测定项下的流动相甲醇-2%冰醋酸(81 : 19)进行实验,但采用该配比测定时维A酸的出峰时间偏长(约30 min),且峰形宽。经实验发现维A酸对甲醇的比例变化敏感,维A酸在甲醇-2%冰醋酸(90∶10)的条件下,出峰时间可缩短为10 min左右,但维A酸与异维A酸的分离度 < 5;而维A酸在甲醇-2%冰醋酸(85∶15)的条件下,出峰时间为18 min左右,维A酸与异维A酸的分离良好(分离度 > 5),为此确定流动相为甲醇-2%冰醋酸(85∶15)。通过扫描维A酸标准溶液的紫外图谱,发现维A酸在354 nm处有最大吸收,因此选择354 nm为检测波长。
3.3 提取条件的选择
本研究分别以无水乙醇、甲醇和流动相作为溶剂提取样品,发现无水乙醇的提取率明显高于甲醇和流动相。为减小供试品溶液对色谱柱的损坏,使用无水乙醇提取后,可冰浴2 h使软膏基质沉淀,滤过后再用流动相稀释进一步析出杂质,最后用0.22 μm微孔滤膜滤过后进样[7]。此外,对提取时间和温度进行优化,发现60 ℃水浴时充分振摇15 min即可完全提取样品。
3.4 维A酸的稳定性
本研究考察了光线对维A酸稳定性的影响。实验结果表明,维A酸溶液在室温光照条件下极不稳定,1 h含量下降近20%,8 h含量下降超过60%;而维A酸在室温避光条件下,12 h基本稳定,因此实验操作中要注意严格避光。
由于维A酸不稳定,见光易转化为异维A酸,而异维A酸具有较强的致畸作用,因此应对制剂中异维A酸的含量进行限制。参考文献[6]维A酸乳膏中的检查方法,本研究将供试品40 μL进样中异维A酸峰面积限定为不大于对照品溶液(10 μL进样)中异维A酸峰面积的5%,经检查所配制的3批样品均符合规定。
综上,采用HPLC法操作简便,准确性高,重复性好,可作为维A酸软膏中维A酸含量测定及有关物质检查的方法。
[1] 中国人民解放军总后勤卫生部.中国人民解放军医疗机构制剂规范(2002年版)[S].北京:人民军医出版社,2003:166.
[2] 姜武民,黄波.维A酸乳膏(0.025%)含量测定方法的探究[J].实用药物与临床,2011,14(3):229-230.
[3] 吴畏,陈雅,杨征,等.紫外分光光度法测定维 A 酸软膏的含量[J].中国药业,2012,21(2):36-37.
[4] 王福霞,冯文菊,姜武民.HPLC法测定维A酸与异维A酸含量[J].药物分析杂志,2007,27(4):585-587.
[5] 魏立明,宋霞,宋三孔,等.HPLC双波长法同时测定复方维A酸软膏中2主药的含量[J].中国药房,2012,23(8):737-739.
[6] 国家药典委员会.中华人民共和国药典(二部)[S]. 2010年版.北京:中国医药科技出版社,2010:893-894.
[7] 蔡乐,白林,徐风华,等.HPLC法测定薄荷苯酚软膏中苯酚的含量[J].中国药物应用与监测,2014,11(2):82-84.
Determination of tretinoin and its related substance in tretinoin ointment
CAI Le, YAO Lan, BAI Lin(Department of Pharmaceutical Care, PLA General Hospital, Beijing 100853, China)
Objective:To establish the HPLC method for determination of tretinoin and its related substance in tretinoin ointment.Methods:HPLC analysis was carried out on a column of Agilent Zorbax SB C18(250 mm × 4.6 mm, 5 μm) with mobile phase consisted of methanol -2% glacial acetic acid solution (85 : 15), the flow rate was 1.0 mL·min-1, the detection wavelength was set at 354 nm and column temperature was 45 ℃.Results:The separation between tretinoin and other impurities was well under this chromatographic condition, and separating degree of tretinoin and isotretinoin was > 5. The calibration curve of tretinoin was linear in the ranger of 0.934 4 – 18.687 2 μg·mL-1(r = 1.000 0). The average recovery of tretinoin was 99.4% (RSD = 1.44%, n = 9).Conclusion:This method is simple, accurate and feasible, which can be applied as the quantitative method for the determination of tretinoin in tretinoin ointment.
HPLC; Tretinoin ointment; Content determination; Tretinoin; Isotretinoin
R917
A
1672 – 8157(2015)03 – 0144 – 03
2014-09-08
2015-01-20)
军队医疗机构制剂标准提高科研专项重点课题:半固体制剂标准提高系列研究(13ZJZ05)
白林,女,主任药师,研究方向:药物分析、医院药学。E-mail:bailin301@126.com
蔡乐,男,主管药师,主要从事药物分析、医院药学工作。E-mail:caile15009@163.com
