一种改良的蛋白质复合物识别方法
2015-01-04栾悉道李学勇
李 彬,栾悉道,王 鑫,李学勇
(长沙大学信息与计算科学系,湖南长沙410022)
近期的实验发展,如酵母双杂交实验[1]、串联亲和纯化[2]以及质谱法[3]已导致经产生了许多高质量、大规模的PPI(蛋白质相互作用)数据.这些数据对于识别蛋白质复合物奠定了基石.蛋白质复合物对于了解细胞的功能组织,从而执行其生物学功能具有非常重要的作用.
然而,由于相关实验技术的局限性和蛋白质相互作用匹配的动态自然性质,从这些高通量的生物实验得到的PPI网络中,相当一部分相互作用包含假阳性[4].研究[5]表明,经过筛选的酵母双杂交数据集中,假阳性相互作用的比例达到50%.这些实验数据中产生的错误将对进一步研究PPI网络带来负面影响.为减少PPI网络中的假阳性,现已提出了几种计算方法预测蛋白质间的相互作用.其中大部分方法借助多种生物信息[6,7],有些方法则依靠统计评分功能[8,9]来计算的蛋白质系谱概要文件的上下文相似性,采用机器学习技术[10]预测蛋白质相互作用网络或使用支持向量机方法[11]构建监督分类来识别相互作用的蛋白质.
为评估高通量蛋白质相互作用的可靠性,现已提出许多计算方法.一些方法被设计用于评估酵母蛋白质相互作用网络的整体误差率[12,13].相互作用数据的比较显得尤为困难,因为他们往往来自不同的条件,呈现出不同的形式.因此,我们采用一些更为复杂的方法来评估单个相互作用的可靠性[14,15].一些基因组信息,如基因??注释,基因表达等已被用于该方法中,而另一些仅使用PPI网络的拓扑结构.
尽管这些计算方法和实验技术取得了长足的进步,我们仍不可能构建一个绝对可靠的PPI网络.所以,对于PPI网络的应用,如复合物的识别、蛋白的功能预测、关键蛋白质的识别以及蛋白质复合物的检测等,相比评估可靠性或减少假阳性,允许假阳性的存在显得更具有必要性和重要性.为了提高预测精度,一些算法考虑到PPI网络的可靠性,并对PPI网络中的相互作用进行加权.例如,CDdistance[16]和 FSWeight[17]两种加权方式,权值根据相互作用的两个蛋白质的共同邻居数量计算.他们已被证实具有较好的性能.即使对于加权PPI网络上运行的这些算法,相互作用的权重通常代表蛋白质相互作用的优先级,描述的一个子图或者蛋白质仍与非加权网络中相同,一些方法引入了加权度.然而,当大量的蛋白质相互作用数据出现时,这些方法不足以得到令人满意的结果.
本文考虑PPI网络中相互作用的可靠性,构建一个非确定性的PPI网络,其中每个相互的可靠性通过一个存在概率表示.我们提出了一种改良的蛋白质复合物识别方法.我们将改良的方法和有代表性的最先进的复合物识别方法作比较.这些方法包括 MCL[18],CMC[16],COACH[19]和 Cluste rONE[20].实验结果表明,我们提出的方法在准确性和统计意义上明显优于这些算法.
1 方法
Gavin[21]等已对复合物组织结构做了深入研究.研究结果显示,一个复合物应该由一个核心组成部分和附件构成.核心蛋白质是高度共表达的,每个附件绑定到核,从而形成具有生物特性的复合物.因此,本文提出的改良的蛋白质复合物识别方法基于核-附件的思想.方法主要包括四个步骤:
(1)计算PPI网络中每组相互作用间的概率及每个蛋白质在其邻居图(即某一蛋白质的所有邻接蛋白质构成的子图,包括这些蛋白质间存在的相互作用)内的支持度,所有蛋白质根据支持度降序排列.
对于任意一组相互作用ei,其相应的存在概率计算如下:
pi=Nic/Nimax其中Nic是相互作用ei的两个蛋白质共同邻居数量,Nimax是两个蛋白质共同邻居数量的最大值.Nimax等于两个蛋白质度的最小值减1.
蛋白质在某一相互作用网络内的支持度定义为:
给定一个蛋白质相互作用网络G=(V,E,P)及蛋白质va∈V,其中V={v1,v2,…,vn}是蛋白质构成的集合,E={e1,e2,…,em}是蛋白质相互作用构成的集合,P={p(e1),p(e2),…,p(em)}是相互作用对应的概率值.蛋白质va在相互作用网络G内的支持度定义为:,其中|V|是G中蛋白质的个数.
在本步骤中,相互作用网络G是某一蛋白质及其所有邻接蛋白质构成的子图,包括这些蛋白质间存在的相互作用.
所有蛋白质在其邻居图内的支持度的平均值Avg_RD被计算,作为步骤3中判定附件蛋白质能否加入核的阈值.
(2)从第一个蛋白质开始,逐渐增加邻居蛋白质来形成具有高凝聚力和低耦合性的候选核.这一过程对所有的蛋白质不断重复直到产生了不重复的核心集.
该方法中,所有蛋白质都享有成为种子的机会.对于每一个选定的种子蛋白质,初始核仅包括种子蛋白质.对于该种子蛋白质的邻接蛋白质,尝试将其加入核,若核的稠密度超过设定的阈值CT,则保留该邻接蛋白质,否则,将该邻接蛋白质从候选核中移除.候选核的稠密度为所有相互作用的概率值总和.种子蛋白质的所有邻居蛋白质都被访问之后,就可以形成一个高内聚的候选核,其内部各个蛋白质间联系比较紧密.
本方法中,每一个核不仅要求内部比较稠密,同时要求核中蛋白质与外部蛋白质间的联系应该比较松散.为此,对于候选核中的蛋白质,若某一蛋白质与核外其他蛋白质的联系相比内部蛋白质间的联系更加紧密,则移除该蛋白质,同时被标记为DISCARD状态.候选核中所有蛋白质均被处理之后所保留的蛋白质便形成一个真正意义上的核.
(3)添加附件蛋白质到核形成蛋白质复合物,其中附件蛋白质与核的期望支持度大于指定的阈值.
只有步骤2中被标记为DISCARD的蛋白质才有可能成为附件蛋白质被添加到核.
对于选定的附件蛋白质,步骤3会将其与所有核做比较,若附件蛋白质在某核内的支持度超过步骤1中得到的阈值Avg_RD,则将附件蛋白质添加到该核中.
(4)计算产生的复合物相互间的重叠率,当重叠率超过指定的阈值时,移除那些具有更低的稠密度或者更小尺寸的复合物.
对于识别的两个复合物A和B,两者的重叠率的计算公式如下[19]:

本方法中重叠率的阈值设为0.8[21].
2 实验结果与分析
本文数据集采用酵母PPI网络,因为酵母PPI网络是所有物种中最完整和可靠的.本文在DIP[22]数据集上运行本方法和其他四种算法:MCL,CMC,COACH和 ClusterONE.DIP数据集发布日期为2012年8月18号,包含4895个蛋白质和21776组蛋白质间的相互作用.为了评估识别的蛋白质复合物,本文采用CYC2008作为已知复合物集,CYC2008包含408个通过生物方法预测得到的复合物.本文主要从准确率、召回率和P-Value值分析对几种算法进行对比分析.为了公平对比,在运行其他四种算法时,相应的参数均按照作者的建议设定为最优值.本次实验设定CT=0.05.
为评估识别方法有效性,将从酵母蛋白质网络中识别出来的复合物与已知复合物进行比较分析.算法识别出来的复合物与已知复合物的匹配程度OS根据公式(1)计算得到.
对于已知复合物数据集中的复合物KC,若识别出的复合物PC与之匹配程度OS(PC,KC)超过给定阈值,则称该已知复合物被标识,一般地,该阈值设置为0.2[19].
算法的准确率(Precision)和召回率(Recall)是用来评估复合物识别方法的两个重要指标.准确率是指识别的复合物中被标识的数量与识别的复合物总量的比值;召回率是指已知复合物中被标识的复合物数量与已知复合物总数的比值.
综合准确率和召回率两个方面,提出了F-Score,它是准确率和召回率的调和平均值,计算公式如(2)所示

表1给出了各种方法识别的复合物的基本信息.其中,Number表示各种识别的复合物总数,MKC是指识别的复合物中被标识的复合物数量,MPC是指已知复合物中被标识的复合物数量,PerfectNumber是完美匹配的复合物数量,即OS=1.
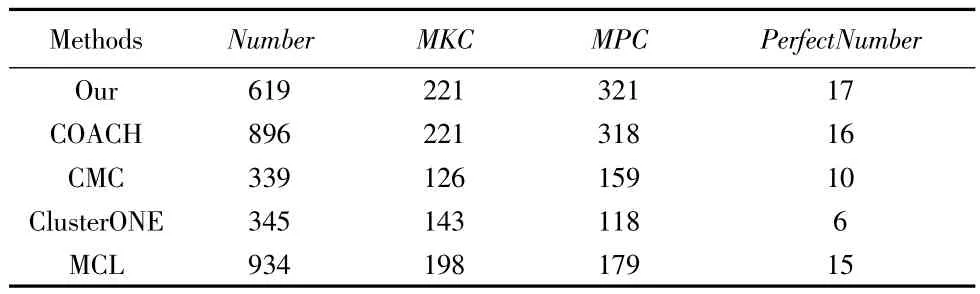
表1 各方法识别的复合物基本信息
从表1可见,本文提出的方法识别的复合物中被标识的数量及完美匹配数量均居榜首.本方法与COACH方法均能使已知复合物中被标识的复合物数量达到最大值(221).对比识别的复合物总数,COACH方法和MCL方法识别的复合物总数均超过本方法.这说明我们的方法具有更高的效率.
图1显示各种方法在DIP数据集中识别的复合物计算的Precision、Recall和F-Score对比分析.
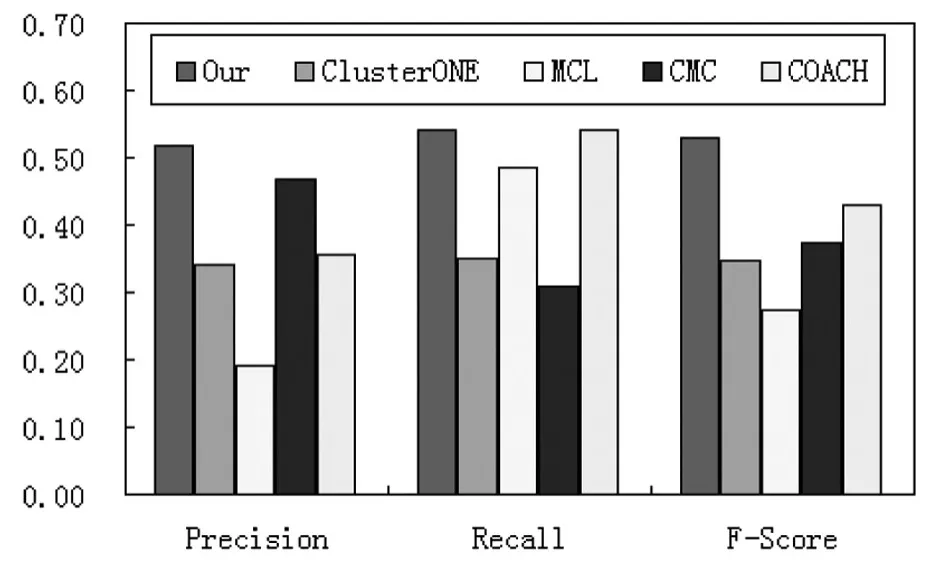
图1 各方法的结果对比分析
图1显示,在DIP数据集中,我们的方法能得到最高的准确率、召回率和F-sccore值,这是我们的方法识别的复合物具有最高的MKC和MPC,而识别的复合物总数并非最多.我们方法的F-Score值分别比COACH、MCL、CMC和ClusterONE 提高了 23.59%,42.31%,53.09%和 92.88%.
本文采用GO功能富集分析评价识别的复合物的统计和生物特性.GO注释是一种非常有效的评估蛋白质相互作用可靠性的资源.GO::TermFinder是一个软件模块集合,决定共享的一组基因的统计学显著特性.为了确定是否任意的GO条目能以一种超过偶然机会的频率注释特定领域的基因列表,GO::TermFinder通过一种超几何分布计算P-value值.如果P-value越小,越接近0,则说明复合物能够随机出现这种功能的概率就越低,当然可能更有生物学意义.同一个复合物内的蛋白质通常具有相同或相似的功能.一般将P-value的最小值对应的功能作为该复合物的主要功能.通过给每个识别的复合物赋予其P-value最小时对应的功能,可以识别未知蛋白质的功能.一般地,若一个复合物的P-value<0.01,则认为这个复合物是显著的.研究指出,显著的复合物数量在识别的复合物总数中所占的比例可以用于评价各个算法的整体性能.另外,P-score值也是另外一种有效的评价手段,其定义如下所示:

表2显示各种方法在DIP数据集上识别的复合物的显著性统计信息.
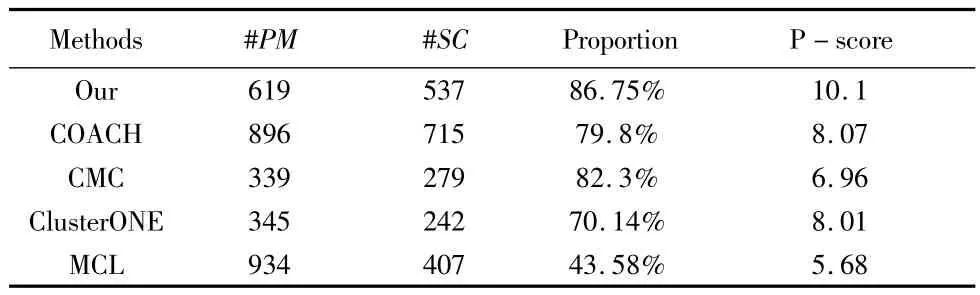
表2 各方法预测的复合物的显著性统计信息
其中,#PM表示算法识别的复合物总数,#SC表示显著的复合物数量,即P-value<0.01的复合物数量.表2显示,我们的方法识别的复合物具有最高的显著性复合物比例和最大的P-score值.我们方法的 P-score值相比 COACH、CMC、ClusterONE 和 MCL,分别提高了25.15%、45.11%、26.09%和77.82%.这说明我们的方法识别的复合物具有最强生物统计意义.
从表2中,我们还可以发现一个有趣的事实:CMC方法的Proportion高于COACH和ClusterONE方法,然而,CMC的P-score确低于 COACH和 ClusterONE.原因在于,虽然COACH和ClusterONE识别的复合物中显著性复合物的比例不如CMC,但是两种方法得到的显著性复合物具有更小的P-value值.

表3 方法识别的复合物的实例
表3是识别的复合物实例.OS表示复合物匹配率,#SC表示正确匹配的蛋白质个数,第四列列举了组成复合物的所有蛋白质,其中,加粗部分表示被匹配的蛋白质.
3 参数分析
步骤(2)中,为了对候选核进行筛选,本文引入自定义参数CT描述子图的稠密度.图2显示了CT取不同值,F-score变化情况.
从图2可以看出,当CT=0.1时,算法得到最高的FScore值.综合考虑完全匹配的复合物数量和F-Score值,本文设定 CT=0.05.

图2 参数CT的影响
4 结论
基于蛋白质间的相互作用存在不可靠性,本文构建一个非确定蛋白质相互作用网络,相互作用的可靠性通过概率值表示,从而尽量消除假阳性或者降低假阳性带来的负面.提出了一种改良的蛋白质复合物识别方法.通过准确率、召回率和P-value几个方面对比分析改良的方法和几种经典的复合物识别方法.实验结果表明,提出的改良方法具有更高的预测准确率,算法识别的复合物具有更强的生物统计特性.
[1]Ito T,Chiba T,Ozawa R,et al.A comprehensive two-hybrid analysis to explore the yeast protein interactome[J].PNAS,2001,(8):4569-4574.
[2]Rigaut G,Shevchenko A,Rutz B,et al.A generic protein purification method for protein complex characterization and proteome exploration[J].Nature Biotechnology,1999,(10):1030-1032.
[3]Ho Y,Gruhler A,Heilbut A,et al.Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry[J].Nature,2002,(6868):180-183.
[4]Mrowka R,Patzak A,Herzel H.Is there a bias in proteome research?[J].Genome research,2001,(12):1971-1973.
[5]Mering C V,Krause R,Snel B,et al.Comparative assessment of large-scale data sets of protein-protein interactions[J].Nature,2002,(6887):399-403.
[6]Tsoka S,Ouzounis C A.Prediction of protein interactions:metabolic enzymes are frequently involved in gene fusion[J].Nature Genetics,2000,(2):141-142.
[7]Wojcik J,Schchter V.Protein– protein interaction map inference using interacting domain profile pairs[J].Bioinformatics,2001,(1):296-305.
[8]Yamada T,Kanehisa M,Goto S.Extraction of phylogenetic network modules from the metabolic network[J].BMC Bioinformatics,2006,(1):130.
[9]Wu J,Kasif S,DeLisi C.Identification of functional links between genes using phylogenetic profiles[J].Bioinformatics,2003,(12):1524-1530.
[10]Albert I,Albert R.Identification of functional links between genes using phylogenetic profiles[J].Bioinformatics,2004,(12):3346-3352.
[11]Lo S,Cai C,Chen Y,et al.Effect of training datasets on support vector machine prediction of protein ‐ protein interactions[J].Proteomics,2005,(4):876-884.
[12]Deane C M,Salwiński L,Xenarios I,et al.:Protein interactions two methods for assessment of the reliability of high throughput observations[J].Molecular& Cellular Proteomics,2002,(5):349-356.
[13]D'haeseleer P,Church G M.Estimating and improving protein interaction error rates[A].Proceedings of Computational Systems Bioinformatics Conference[C].IEEE,2004:216-223.
[14]Gilchrist M A,Salter L A,Wagner A.A statistical framework for combining and interpreting proteomic datasets[J].Bioinformatics,2004,(5):689-700.
[15]Mering V C,Krause R,Snel B,et al.Comparative assessment of large-scale data sets of protein– protein interactions[J].Nature,2002,(6887):399-403.
[16]Liu G,Wong L,Chua H N.Complex discovery from weighted PPI networks[J].Bioinformatics,2009,(15):1891-1897.
[17]Chua H N,Sung W K,Wong L.Exploiting indirect neighbours and topological weight to predict protein function from protein–protein interactions[J].Bioinformatics,2006,(3):1623-1630.
[18]Enright A J,Dongen S V,Ouzounis C A.An efficient algorithm for large-scale detection of protein families[J].Nucleic Acids Research,2002,(7):1575-1584.
[19]Wu M,Li X,.Chee-Keong K,et al.A core-attachment based method to detect protein complexes in ppi networks[J].BMC Bioinformatics,2009,(1):169.
[20]Nepusz T,Yu H,Paccanaro A.Detecting overlapping protein complexes in protein-protein interaction networks[J].Nature Methods,2012,(5):471-475.
[21]Gavin A,Aloy P,Grandi P,et al.Proteome survey reveals modularity of the yeast cell machinery[J].Nature,2006,(7084):631-636.
[22]Xenarios I,Salwinski L,Duan X J,et al.DIP,the database of interacting proteins:A research tool for studying cellular networks of protein interactions[J].Nucleic acids research,2002,(1):303-305.
