SU11274逆转肝细胞生长因子诱导不同EGFR基因型非小细胞肺癌细胞株对吉非替尼耐药
2015-01-04严春花玄香兰张佳安昌善
严春花,玄香兰,张佳,安昌善
1.延边大学附属医院呼吸内科,吉林 延吉133000;
2.延边第二人民医院呼吸内科,吉林 延吉133000
SU11274逆转肝细胞生长因子诱导不同EGFR基因型非小细胞肺癌细胞株对吉非替尼耐药
严春花1,玄香兰2,张佳1,安昌善1
1.延边大学附属医院呼吸内科,吉林 延吉133000;
2.延边第二人民医院呼吸内科,吉林 延吉133000
背景与目的:肝细胞生长因子(hepatocyte growth factor,HGF)诱导敏感非小细胞肺癌(nonsmall cell lung cancer,NSCLC)细胞对表皮生长因子受体酪氨酸激酶抑制剂(epidermal growth factor receptor-tyrosine kinase inhibitor,EGFR-TKI)耐药,其机制与c-Met激活有关。本研究探讨c-Met抑制剂SU11274逆转HGF诱导的不同EGFR基因型NSCLC细胞株对吉非替尼耐药及逆转耐药机制。方法:选择人NSCLC细胞株PC9(EGFR突变型)、H292(EGFR野生型)和A549(EGFR野生型),应用吉非替尼和SU11274单独或联合作用于HGF诱导的细胞株。实验分为6组:C组(不加药对照组)、H组(HGF处理组)、G组(吉非替尼处理组)、S组(SU11274处理组)、HG组(HGF+吉非替尼处理组)和HGS组(HGF+吉非替尼+SU11274处理组)。MTT法检测对细胞增殖的影响,流式细胞术检测细胞凋亡的影响;应用蛋白质印迹法(Western blot)检测细胞中c-Met及其下游通道Stat3、Akt和Erk1/2蛋白表达水平。结果:吉非替尼对3种细胞的生长抑制作用均呈浓度依赖性,HGF处理能够缓解吉非替尼的增殖抑制作用(P<0.05);不同浓度吉非替尼联合SU11274作用于HGF诱导细胞时,3种细胞株存活率比吉非替尼单独作用于HGF诱导细胞时明显降低(P<0.05);HGS组的细胞凋亡比HG组明显增加(P<0.05);HGS组的c-Met、Stat3、Akt和Erk1/2活化蛋白量比HG组明显减少。结论:c-Met抑制剂SU11274可逆转HGF诱导的不同EGFR基因型NSCLC细胞株对吉非替尼耐药,其机制可能与抑制HGF活化的c-Met及其下游通道蛋白表达有关。
肝细胞生长因子;吉非替尼;耐药:SU11274;非小细胞肺癌
目前,分子靶向药物表皮生长因子受体酪氨酸激酶抑制剂(epidermal growth facter receptortyrosine kinase inhibitor,EGFR-TKI)逐渐成为治疗非小细胞肺癌(non-small cell lung cancer,NSCLC)的重要手段。EGFR-TKI有效率与EGFR基因型有很大的相关性[1-2],存在原发或获得性耐药,开始对EGFR-TKI疗效明显的患者一段时间后逐渐出现获得性耐药[3]。HGF诱导NSCLC细胞对吉非替尼耐药,且与c-Met激活有关[4-6]。有研究报道,c-Met抑制剂(E7050、PF-2341066)可逆转HGF诱导的EGFR突变型NSCLC细胞株对吉非替尼耐药[7-8]。SU11274是选择性的c-Met抑制剂。本研究选择EGFR不同基因型人NSCLC细胞(EGFR突变型和野生型),用SU11274作用于HGF诱导对吉非替尼耐药的NSCLC细胞,检测其对细胞增殖、凋亡以及c-Met信号通道蛋白的影响,探讨SU11274是否逆转HGF诱导的不同EGFR基因型NSCLC细胞对吉非替尼耐药及逆转耐药机制,为临床提供联合用药的理论依据。
1 材料和方法
1.1 实验材料
选择人NSCLC细胞株PC9(EGFR突变型,敏感株)、H292(EGFR野生型,敏感株)和A 5 4 9(E G F R野生型,原发性耐药株),均由上海市肺科医院中心实验室提供;DMEM培养液及新生牛血清购自美国Hyclone公司;吉非替尼原料购自济南汇丰达化工有限公司;HGF购自美国Humanzyme公司;SU11274购自美国Sigma公司;MTT粉购自美国Amresco公司;Annexin Ⅴ-FITC凋亡检测试剂盒购自美国BD PharMingen公司。兔抗人p-Met(Tyr1349)、c-Met、p-Akt(Ser473)、Akt、Erk1、p-Stat3(Ser727)、Stat3和GAPDH抗体均购自美国Epitomics公司,p-Erk1/2(Tyr202/Y204)购自美国Cell Signaling Technology公司,辣根过氧化物酶标记的羊抗兔购自美国Jection公司;NC膜购自美国Whatman公司;ECL化学发光试剂购自美国Thermo公司。其它试剂均为国产分析纯。
1.2 细胞培养及药物配制
分别将PC9、H292和A549细胞常规培养于含10%新生牛血清的DMEM培养液中,置于CO2体积分数为5%、37 ℃恒温细胞培养箱中温育,每3~4 d换液传代1次。HGF用含0.1%BSA的蒸馏水稀释成500 μg/mL的母液。吉非替尼原料用DMSO溶解稀释成浓度为100 mmol/L的母液,用药时DMSO终浓度<0.1%。SU11274用DMSO溶解稀释成浓度为100 mmol/L的母液,用药时DMSO终浓度<0.1%。
1.3 MTT法检测细胞增殖
取100 μL含细胞数为1×10³个的细胞悬浮液接种于96孔板,每组设5个复孔。细胞贴壁后,每孔内加入40 ng/mL HGF和(或)浓度为0、0.01、0.04、0.1、0.4、1、4和10 μmol/L的吉非替尼和(或)最小SU11274浓度。72 h后,每孔加入20 μL MTT(5 mg/mL),放入细胞培养箱中温育。4 h后,离心、弃上清液,每孔加入200 μL DMSO,摇床上混匀约30 min至结晶完全溶解,用酶标仪测量波长530 nm时吸光度(A)值。细胞存活率(%)=(A实验组均值-A空白组均值)/(A对照组均值-A空白组均值)×100%。实验重复3次。用细胞存活率做出量效曲线,用作图法分析得出1种药物对不同细胞的IC50。
1.4 流式细胞仪检测细胞凋亡
取5×105个对数生长期细胞接种于6孔板。贴壁后弃原培养液,加入40 ng/mL HGF和(或)1 μmol/L吉非替尼和(或)最小SU11274浓度。48 h后,胰酶消化并收集全部细胞到5 mL试管中,离心、弃上清液,0.9%NaCl溶液洗涤1次。加入1×Binding Buffer调整细胞浓度为1×106个/mL,取100 μL(1×105个细胞)到新的5 mL试管中。各管内加入5 μL FITC和5 μL PI,室温、避光15 min。上机前加入400 μL 1×Binding Buffer,在1 h内进行检测。实验重复3次。
1.5 蛋白质印迹法(Western blot)检测蛋白表达水平
取5×105个对数生长期的细胞接种于6 mm2细胞培养皿,细胞贴壁后加入40 ng/mL HGF和浓度分别为0、0.5、1、2.5、5和10 μmol/L的SU11274。24 h后,冰上裂解细胞提取总蛋白,用BCA法定量,取30~40 μg蛋白经8%~10% SDSPAGE电泳分离后转印至NC膜上,用5%脱脂奶粉封闭1 h,加入一抗温育4 ℃过夜,TBST洗膜10 min、3次后二抗室温摇床温育1 h,TBST洗膜5 min、5次后ECL化学发光试剂显色、曝光成像。ImageJ2x软件测定各条带灰度值。选择其中SU11274最小有效抑制浓度后,每种细胞实验分6组:C组(对照组)、H组(HGF处理组)、 S组(SU11274处理组)、G组(吉非替尼处理组)、HG组(HGF+吉非替尼处理组)和HGS组(HGF+吉非替尼+SU11274处理组)。加入40 ng/mL HGF和(或)1 μmol/L吉非替尼和(或)最小SU11274浓度,检测各组细胞内c-Met、p-Met、Stat3、p-Stat3、Akt、p-Akt、Erk1/2和p-Erk1/2蛋白的表达水平。
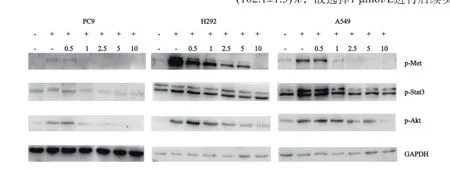
图1 不同浓度SU11274作用后c-Met下游通道蛋白磷酸化情况Fig. 1 The phosphorylation of c-Met downstreams signaling pathway proteins after treating with different concentrations of SU11274
1.6 统计学处理
采用SPSS 17.0统计学软件对结果进行统计分析。计量资料用±s 表示,2组间比较采用t检验,P<0.05为差异有统计学意义。
2 结 果
2.1 SU11274单独作用对HGF诱导的细胞中c-Met及其下游信号蛋白表达的影响
不同浓度SU11274作用于HGF诱导的细胞后,Western blot检测结果显示,p-Met、p-Stat3和p-Akt蛋白表达有不同程度受到抑制。SU11274浓度为1 μmol/L时HGF刺激3种细胞被激活的c-Met、p-Stat3和p-Akt蛋白表达均开始受到抑制(图1)。而1 μmol/L SU11274对PC9、H292和A549细胞生长几乎没有抑制作用,其细胞存活率分别为(96.8±3.2)%、(95.9±2.5)%和(102.1±1.3)%,故选择1 μmol/L进行后续实验。
2.2 吉非替尼单独作用和联合SU11274对HGF诱导的细胞生长抑制的影响
吉非替尼以不同浓度单独作用于PC9、H292和A549细胞72 h后,MTT法检测结果显示,均产生细胞生长抑制作用,且呈浓度依赖性。吉非替尼作用于HGF诱导的细胞后,其IC50值比非HGF诱导组明显增高,与SU11274联合作用于HGF诱导的细胞后IC50值明显降低(P<0.05,图2)。在吉非替尼相同浓度的条件下,HGF诱导组细胞存活率比非诱导组明显提高,差异有统计学意义(P<0.05,图2)。

图2 吉非替尼作用于HGF单用或联合SU11274处理的PC9、H292、A549细胞株药物浓度-存活率曲线Fig. 2 The concentration-survival curve of gefitinib and HGF treated PC9, H292, A549 cells with or without SU11274
1 μmol/L SU11274联合不同浓度吉非替尼(0.01、0.04、0.1、0.4、1、4和10 μmol/L)作用于HGF诱导的细胞72 h后,在相同药物浓度下细胞存活率比单独不同浓度吉非替尼(0.01、0.04、0.1、0.4、1、4和10 μmol/L)作用于HGF诱导的细胞存活率明显降低,差异有统计学意义(P<0.05,图2)。
2.3 吉非替尼单独作用和联合SU11274对HGF诱导的细胞凋亡的影响
用1μmol/L吉非替尼作用于PC9、H292和A549细胞72 h后,细胞凋亡率分别为(4.7±2.0)%、(31.8±10.4)%和(10.8±3.6)%。用1 μmol/L吉非替尼单独作用于HGF诱导的细胞72 h后,细胞凋亡率分别为(4.4±1.7)%、(21.5±8.2)%和(3.1±1.4)%。除了PC9细胞外,HGF诱导组的凋亡率比非HGF诱导组显著减少(P<0.05)。SU11274(1 μmol/L)联合吉非替尼(1 μmol/L)作用于HGF诱导的细胞72 h后,细胞凋亡率比单用吉非替尼作用于HGF诱导细胞后的凋亡率显著升高(P<0.05,表1)。
2.4 吉非替尼单独或联合SU11274对HGF诱导的细胞中c-Met及其下游通道蛋白的影响
Western blot检测结果显示,吉非替尼单独作用于诱导前细胞时,细胞中p-Stat3、p-Akt和p-Erk1/2蛋白表达水平均明显下降。吉非替尼单独作用于HGF诱导的细胞时,细胞中p-Met、p-Stat3、p-Akt和p-Erk1/2蛋白表达水平均明显升高。当SU11274和吉非替尼联合作用于HGF诱导的细胞时,细胞中p-Met、p-Stat3、p-Akt和p-Erk1/2蛋白表达水平均明显下降(图3)。

表1 吉非替尼单用或联合SU11274对HGF诱导的细胞凋亡率Tab. 1 The effects of gefitinib with or without SU11274 on the apoptosis induced by HGF
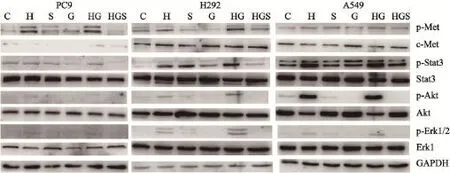
图3 吉非替尼单用或联合SU11274作用对HGF诱导的c-Met及其下游通道蛋白表达的影响Fig. 3 The expressions of HGF-induced c-Met and downstream signaling pathway proteins affected by gefitinib alone or accompanied with SU11274
3 讨 论
EGFR是一种跨膜酪氨酸激酶受体,是人类表皮生长因子受体家族中的一员。EGFR基因与配体结合后,EGFR基因在细胞内区的酪氨酸发生自身磷酸化,活化下游信号通道,调控上皮来源细胞的增殖、侵袭及迁移。吉非替尼是小分子EGFR-TKI,阻止EGFR自身磷酸化,阻断EGFR介导的下游通道活性,表现为抑制肿瘤细胞生长、迁移、侵袭和肿瘤血管生成等。EGFR突变与EGFR-TKI有效率有很大的相关性,存在原发性或获得性耐药。EGFR基因2次突变(T790M)[9]和MET基因扩增[10]是EGFR-TKI耐药的两大主要分子机制,其它可能的机制有胰岛素样生长因子1受体(IGF-1R)过表达[11]、蛋白酪氨酸磷酸酶基因(PTEN)缺失[12]、HGF高表达[13-14]等。HGF/c-Met信号通路参与EGFRTKI获得性耐药[4-5]。
HGF属于成纤维细胞的衍生因子,与特异性c-Met受体结合,使其发生自身磷酸化,激活细胞内信号途径,促进多种组织细胞增生分裂、细胞运动和血管生成。在各种恶性肿瘤中发现异常的HGF/c-Met信号通道,且与不良预后相关。Senguta等[15]测定高侵袭状态肺癌患者血清中HGF含量明显升高,血清HGF含量升高可能与EGFR-TKI耐药有关。目前,c-Met抑制剂主要有PF02341066、GW2974和SU11274等。SU11274可减少T790M-EGFR型NSCLC细胞对EGFR-TKI耐药[16-17]。本研究选择的PC9细胞具有EGFR基因突变型敏感株,H292细胞为EGFR基因野生型敏感株,A549细胞为EGFR基因野生型耐药株,从IC50值可以看出其耐药性。吉非替尼抑制PC9、H292、A549细胞增殖作用均呈浓度依赖性,HGF诱导后IC50显著升高,说明HGF明显提高吉非替尼抑制细胞增殖的IC50,使3种肺癌细胞存活率升高,增加了耐药性。吉非替尼促进细胞凋亡,HGF诱导后则明显降低其促进细胞凋亡作用。
细胞除了表达EGFR外同时还表达其它含酪氨酸激酶活性的跨膜受体,称之为EGFR旁路酪氨酸激酶信号。c-Met属于受体酪氨酸激酶家族的一员。c-Met与其配体HGF结合,激活细胞内MAPKs、PI3K/Akt和c-Src/FAK、STAT信号通路。本研究中,吉非替尼对PC9、H292、A549的生长抑制作用呈浓度依赖性,HGF诱导后吉非替尼抑制细胞的生长曲线往右移。HGF诱导后吉非替尼对PC9、H292、A549细胞的IC50值明显升高,使不同基因型肺癌细胞存活率升高。40 ng/mL HGF和不同浓度吉非替尼及SU11274 (1 μmol/L)作用后,PC9、H292和A549细胞的存活率恢复到原来的水平。在EGFR野生型细胞中,HGF能明显减少吉非替尼对肺癌细胞的凋亡,而在EGFR突变型细胞中,HGF不能明显减少吉非替尼对肺癌细胞的凋亡。在SU11274联合作用下,吉非替尼对HGF诱导3种肺癌细胞的凋亡率显著提高。
HGF与c-Met结合后c-Met发生自身磷酸化,激活Met下游通道(MAPK、PI3K/Akt、c-Src/ FAK和STAT等)。c-Met抑制剂和EGFR-TKI联合用药逆转HGF诱导的EGFR突变型NSCLC细胞株对EGFR-TKI耐药,其机制与抑制c-Met下游通道激活有关[3-4]。本研究结果显示,吉非替尼在无HGF刺激下明显抑制PC9、H292和A549细胞内Akt、Stat3及Erk1/2的活化,但在HGF诱导下不能抑制PC9、H292和A549细胞内c-Met、Akt、Stat3和Erk1/2活化,同时检测的非磷酸化的c-Met、Akt、Stat3和Erk1/2总量无明显变化。说明c-Met及其下游通道蛋白磷酸化参与了HGF诱导不同EGFR基因型NSCLC对吉非替尼耐药机制。
本研究中,在HGF诱导的PC9、H292和A549细胞凋亡,吉非替尼联合SU11274组比单用吉非替尼组HGF激活的c-Met及其下游通道蛋白c-Met、Akt、Stat3和Erk1/2的磷酸化表达减少。在吉非替尼作用下,PC9、H292和A549细胞中HGF诱导激活的c-Met及其下游通道蛋白c-Met、Akt、Stat3和Erk1/2被SU11274抑制。提示c-Met抑制剂可逆转HGF诱导不同EGFR基因型NSCLC细胞株对吉非替尼耐药,其机制与逆转c-Met及下游通道c-Met、Akt、Stat3和Erk1/2的激活有关。联合应用EGFR-TKI与HGF拮抗剂或c-Met抑制剂对分泌HGF或表达c-Met及EGFR-TKI耐药的NSCLC患者可能更加有效,其临床疗效令人期待。
[1] PAEZ J G, JANNE P A, TEE J C, et a1. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy[J]. Science, 2004, 304(5676): 1497.
[2] FUKUOKA M, YANO S, GIACCONE G, et al. Multiinstitutional randomized phase Ⅱ trial of gefitinib for previously treated patients with advanced non-small cell lung cancer (The IDEAL 1 Trial)[J]. J Clin Oncol, 2003, 21(12): 2237-2246.
[3] MITSUDOMI T, YATABE Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer[J]. Cancer Sci, 2007, 98(12): 1817-1824.
[4] 玄香兰, 安昌善, 周彩存. 肝细胞生长因子诱导敏感非小细胞肺癌细胞对吉非替尼耐药及机制的研究[J]. 中国肺癌杂志, 2013, 16(1): 1-6.
[5] 玄香兰, 安昌善, 周彩存. c-Met信号通道参与HGF诱导不同基因型非小细胞肺癌细胞株对吉非替尼耐药[J]. 中国肺癌杂志, 2013, 16(9): 464-469.
[6] YANO S, WANG W, LI Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor–activating mutations[J]. Cancer Res, 2008, 68(22): 9479-9487.
[7] WANG W, LI Q, TAKEUCHI S, et al. Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor-induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer[J]. Clin Cancer Res, 2012, 18(6): 1663-1671.
[8] CHEN X, ZHOU J Y, ZHAO J, et al. Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small cell lung cancer cells[J]. Anticancer Drugs, 2013, 24(10): 1039-1046.
[9] KOBAYASHI S, BOGGON T J, DAYARAM T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib[J]. N Engl J Med, 2005, 352: 786-792.
[10] ENGELMAN J A, ZEJNULLAHU K, MITSUDOMI T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling[J]. Science, 2007, 316: 1039-1043.
[11] GUALBERTO A, POLLAK M. Emerging role of insulin-like growth factor receptor inhibitor in oncology early clinical trial results and future directions[J]. Oncogene, 2009, 289(34): 3009-3021.
[12] BIANCO R, SHIN I, RITTER C A , et al. Loss of PTEN/ MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors[J]. Oncogene, 2003, 22 (18): 2812-2822.
[13] WANG W, LI Q, YAMADA T, et al. Cross talk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors[J]. Clin Cancer Res, 2009, 15 (21): 6630-6638.
[14] GUO A, VILLEN J, KORNHAUSER J, et al. Signaling network assembled by oncogenic EGFR and c-Met[J]. PNAS, 2008, 105(2): 692-697.
[15] SENGUTA S, GHERADI E, SELLERA L A, et al. Hepatocyte growth factor/scater factor can induce angiogenesis independently of vascular endothelial growth factor[J]. Arterioscler Thromb Vasc Biol, 2003, 23(1): 69-75.
[16] TANG Z, DU R, JIANG S, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer[J]. Br J Cancer, 2008, 99(6): 911-922.
[17] CHEN G, NOOR A, KRONENBERGER P, et al. Synergistic effect of afatinib with su11274 in non-small cell lung cancer cells resistant to gefitinib or erlotinib[J]. PLoS One, 2013, 8(3): e59708.
SU11274 reverse gefitinib resistance induced by hepatocyte growth factor in different EGFR gene type of non-small cell lung cancer cells
YAN Chunhua1, XUAN Xianglan2, ZHANG Jia1, AN Changshan1(1.Department of Respiratory Disease, Yanbian University Hospital, Yanji Jilin 133000, China; 2. Department of Respiratory Disease, Yanbian Second People’s Hospital, Yanji Jilin 133000, China)
AN Changshan E-mail: cs_an2003@aliyun.com
Background and purpose:Hepatocyte growth factor (HGF) induce epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) resistance in non-small cell lung cancer (NSCLC) cells, the mechanism might be related with activation of c-Met. The present study aimed to explore whether c-Met inhibitor SU11274 reverse gefitinib resistance induced by HGF in different EGFR gene types of NSCLC.Methods:PC9 (EGFR-activating mutant), H292 (EGFR-wild type) and A549 (EGFR-wiled) were chosen. The experiments were divided into 6 groups: C group (control), H group (HGF), G group (geftinib), S group (SU11274), GH group (geftinib+HGF), GSH group (geftinib+SU11274+HGF). The cell survival was measured by MTT assay; the cell apoptosis was measured by fow cytometry (FCM); the expressions of c-Met, Stat3, Akt and Erk1/2 protein were examined by Western blot.Results:Gefitinib inhibited cell growth of 3 cells lines in a dose-dependent manner, and treating with HGF could relieveinhibition of cell growth caused by geftinib. The cell survival when treating the HGF-induced cell lines with defferent concentration of geftinib combined with SU11274 was signifcantly decreased than that when treating HGF-induced cell lines with geftinib alone. In 3 cell lines, the apoptosis rate in HGS group was higher than that in HG group (P<0.05). In three cells lines, the p-Met, p-Stat3, p-Akt and p-Erk1/2 expressions in HGS group were lower than that in HG group (P<0.05).Conclusion:SU11274 reversed geftinib resistance induced by HGF in different EGFR gene types of NSCLC cells, the mechanism might be related with inhibiting the HGF-induced activation of c-Met and its downstream signaling pathway.
Hepatocyte growth factor; Geftinib; Resistance; SU11274; Non-small cell lung cancer
10.3969/j.issn.1007-3969.2015.02.004
R735.7
A
1007-3639(2015)02-0099-06
国家自然科学基金项目(81160291)。
安昌善 E-mail:cs_an2003@aliyun.com
