2-甲基噻吩与NO3反应机理的理论研究
2014-12-31王渭娜卢天宇王文亮
陈 龙,王渭娜,卢天宇,王文亮
(陕西省大分子科学重点实验室,陕西师范大学 化学化工学院,陕西 西安 710119)
噻吩及其衍生物是含硫的有机杂环化合物,其中噻吩、2-甲基噻吩、3-甲基噻吩和2,5-二甲基噻吩是医药制造、松香合成等工业生产的重要原料.在大气中,它们来源于生物体释放或植被、煤炭、生物体的燃烧等人为排放[1-5].大气中的噻吩及其衍生物主要通过与OH、NO3、O3自由基发生氧化反应得以降解[6-7].光照条件下,OH 与大量的杂环有机物发生反应,在夜间,OH不易与杂环有机物发生反应.而NO3见光易分解,夜间浓度高,可大量参与大气化学反应[8-12].由此可见,NO3与杂环有机物反应是夜间大气进行的主要反应,对其进行研究有重要的现实意义.
1 计算方法
本文采用密度泛函理论(Density Functional Theory,DFT)中的 B3LYP[13]方法对2-甲基噻吩与NO3反应机理进行研究.在B3LYP/6-31+G(d,p)水平上对反应路径上各驻点(反应物、中间体、过渡态、复合物和产物)进行几何构型优化,并在相同水平上进行内禀反应坐标(IRC)分析[14-15],以证实反应物、中间体、过渡态和产物的相关性.为了获得更加精确的反应最小能量路径(VMEP(s))信息,采用B3LYP/6-311++G(3df,2pd)方法进行单点能计算.以上全部工作用Gaussian 03程序[16]在陕西师范大学理论与计算化学研究室的宝德高性能服务器上完成.
采用传统过渡态理论(TST)计算各温度下双分子反应的速率常数,通过 Wigner校正[17-18]后的速率常数(kn)如公式(1)所示.
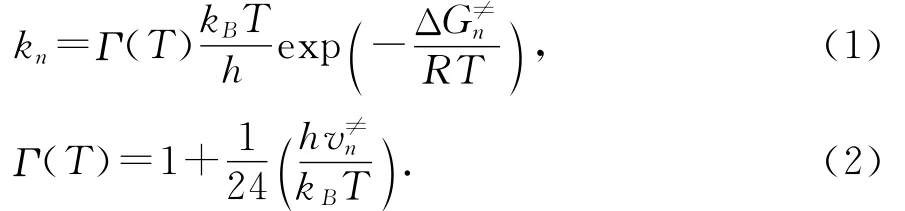
式中:Γ(T)为 Wigner校正因子,kB是玻尔兹曼常数,h是普朗克常数,R是摩尔气体常数,v≠n为反应过渡态虚频的绝对值,ΔG≠n是考虑了零点能校正后的速控步骤活化吉布斯自由能.以上所有工作是应用VKLab程序包[19]完成的.
2 结果与讨论
已有文献证实噻吩与NO3反应存在3种可能的反应机理[4,12],分别为抽氢、SN2取代和加成-消去反应.为了便于与已知的反应通道进行比较,本文主要考察2-甲基噻吩与NO3反应的抽氢、SN2取代和加成-消去3种反应类型,共10条反应通道,如图1所示.图2列出在B3LYP/6-31+G(d,p)水平上优化的各反应物、中间体、过渡态和产物的几何构型.图3绘出标题反应在 B3LYP/6-311++G(3df,2pd)//B3LYP/6-31+G(d,p)+ZPE水平上的势能剖面图.表1列出不同驻点物种的电子结构能(E)、零点能(ZPE)、总能量(ET)、相对能量(ER)和活化能垒(E#a).表2列出298.15K下各通道速控步的吉布斯自由能(ΔG≠n)和反应速率常数(kn).
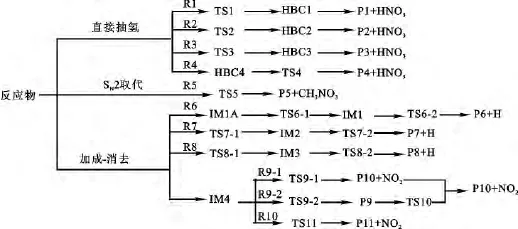
图1 标题反应路径图Fig.1 Main reaction paths of the title reactions
2.1 反应机理
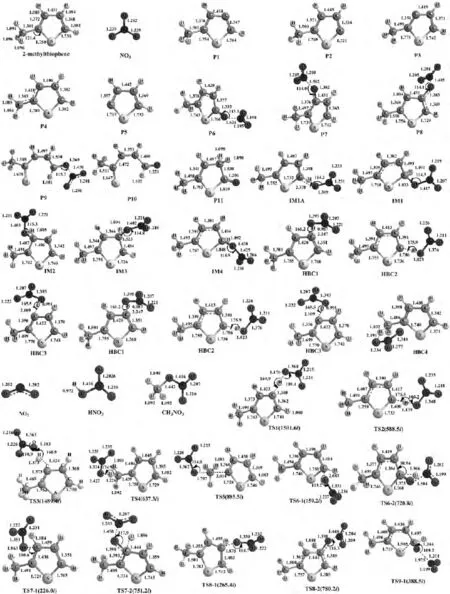
图2 标题反应在B3LYP/6-31+G(d,p)水平上优化所得的各反应物、产物、中间体、复合物和过渡态的几何构型Fig.2 Geometric structures of the transition states,intermediates,complexes,reactants and products of the title reaction optimized at the B3LYP/6-31+G (d,p)level
表1 不同驻点物种的结构能(E)、零点能(ZPE)、总能量(ET)、相对能量(ER)和活化能垒()Tab.1 Electronic structure energies(E),zero point energy(ZPE),the total energy(ET),the relative energy(ER)and activation free energy()for the species involved in the title reactions
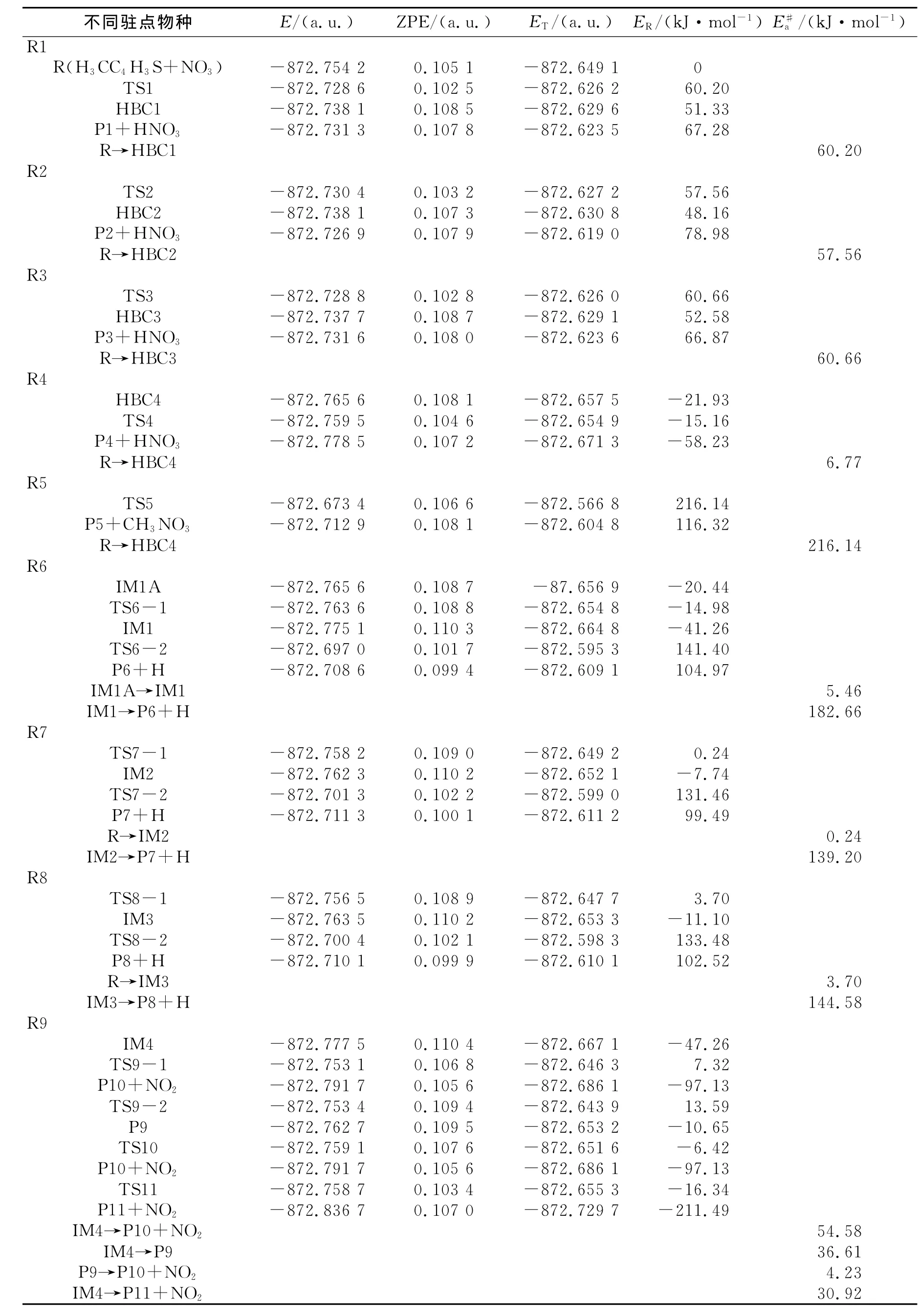
表1 不同驻点物种的结构能(E)、零点能(ZPE)、总能量(ET)、相对能量(ER)和活化能垒()Tab.1 Electronic structure energies(E),zero point energy(ZPE),the total energy(ET),the relative energy(ER)and activation free energy()for the species involved in the title reactions
不同驻点物种E/(a.u.)ZPE/(a.u.)ET/(a.u.)ER/(kJ·mol-1)E#a/(kJ·mol-1)R1 60.20 R2 TS2 -872.730 4 0.103 2 -872.627 2 57.56 HBC2 -872.738 1 0.107 3 -872.630 8 48.16 P2+HNO3 -872.726 9 0.107 9 -872.619 0 78.98 R→HBC2 57.56 R3 TS3 -872.728 8 0.102 8 -872.626 0 60.66 HBC3 -872.737 7 0.108 7 -872.629 1 52.58 P3+HNO3 -872.731 6 0.108 0 -872.623 6 66.87 R→HBC3 60.66 R4 HBC4 -872.765 6 0.108 1 -872.657 5 -21.93 TS4 -872.759 5 0.104 6 -872.654 9 -15.16 P4+HNO3 -872.778 5 0.107 2 -872.671 3 -58.23 R→HBC4 6.77 R5 TS5 -872.673 4 0.106 6 -872.566 8 216.14 P5+CH3NO3 -872.712 9 0.108 1 -872.604 8 116.32 R→HBC4 216.14 R6 IM1A -872.765 6 0.108 7 -87.656 9 -20.44 TS6-1 -872.763 6 0.108 8 -872.654 8 -14.98 IM1 -872.775 1 0.110 3 -872.664 8 -41.26 TS6-2 -872.697 0 0.101 7 -872.595 3 141.40 P6+H -872.708 6 0.099 4 -872.609 1 104.97 IM1A→IM1 5.46 IM1→P6+H 182.66 R7 TS7-1 -872.758 2 0.109 0 -872.649 2 0.24 IM2 -872.762 3 0.110 2 -872.652 1 -7.74 TS7-2 -872.701 3 0.102 2 -872.599 0 131.46 P7+H -872.711 3 0.100 1 -872.611 2 99.49 R→IM2 0.24 IM2→P7+H 139.20 R8 TS8-1 -872.756 5 0.108 9 -872.647 7 3.70 IM3 -872.763 5 0.110 2 -872.653 3 -11.10 TS8-2 -872.700 4 0.102 1 -872.598 3 133.48 P8+H -872.710 1 0.099 9 -872.610 1 102.52 R→IM3 3.70 IM3→P8+H 144.58 R9 IM4 -872.777 5 0.110 4 -872.667 1 -47.26 TS9-1 -872.753 1 0.106 8 -872.646 3 7.32 P10+NO2 -872.791 7 0.105 6 -872.686 1 -97.13 TS9-2 -872.753 4 0.109 4 -872.643 9 13.59 P9 -872.762 7 0.109 5 -872.653 2 -10.65 TS10 -872.759 1 0.107 6 -872.651 6 -6.42 P10+NO2 -872.791 7 0.105 6 -872.686 1 -97.13 TS11 -872.758 7 0.103 4 -872.655 3 -16.34 P11+NO2 -872.836 7 0.107 0 -872.729 7 -211.49 IM4→P10+NO2 54.58 IM4→P9 36.61 P9→P10+NO2 4.23 IM4→P11+NO2 R(H3CC4H3S+NO3) -872.754 2 0.105 1 -872.649 1 0 TS1 -872.728 6 0.102 5 -872.626 2 60.20 HBC1 -872.738 1 0.108 5 -872.629 6 51.33 P1+HNO3 -872.731 3 0.107 8 -872.623 5 67.28 R→HBC1 30.92
2.1.1 抽氢反应 在抽氢反应中,NO3中的一个O原子抽取2-甲基噻吩环上不同位置的H(噻吩环上的含氢C原子从右向左依次标记为C1、C2和C3),反应标记为R1、R2和R3(如图1所示).另外,2-甲基噻吩分子中甲基上的3个H原子近似等价,因此本文仅考虑抽取其中一个氢的反应,标记为R4.在抽氢反应R1中,从反应物出发,经过渡态TS1转化为产物复合物HBC1.在过渡态TS1中,NO3中的O直接抽取环上的H生成产物C5H6S(P1)+HNO3.此过程的能垒为60.20kJ/mol(如表1和图3所示),且为吸热过程(70.82kJ/mol).R2和 R3与 R1反应过程相似,均由反应物出发,经过渡态TS2和TS3生成产物复合物HBC2和HBC3,所需能垒分别为57.56和 60.66kJ/mol,且均为吸热反应(83.02和70.50kJ/mol).因此,无论是从热力学还是从动力学方面考虑,这3条通道都不易进行.
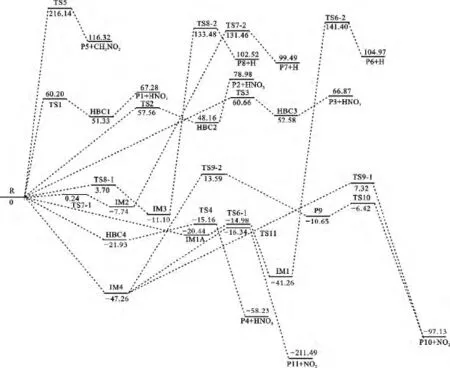
图3 标题反应在B3LYP/6-311++G(3df,2pd)//6-31+G(d,p)+ZPE水平上的势能剖面图Fig.3 Potential energy profile of the title reactions at the B3LYP/6-311++G(3df,2pd)//6-31+G(d,p)+ZPE level(kJ/mol)
NO3抽取甲基上H的反应(R4)与抽取环上氢(R1、R2、R3)有一些不同点:首先 NO3中的 O与甲基上的H形成氢键复合物HBC4,此过程释放出的能量为21.93kJ/mol;再由复合物HBC4经过渡态TS4(对应的虚频值为637.3icm-1)向产物 P4+HNO3转化,伴随着C-H键的断裂和O-H的生成,此过程仅需越过6.77kJ/mol的能垒,且是强放热反应,形成类似苄基结构稳定自由基产物C5H6S(P4).因此,无论是热力学还是动力学都容易进行,即抽取甲基上的H为优势反应通道.
2.1.2 SN2取代反应 NO3可以分别从异侧和同侧取代噻吩环形成CH3NO3,但从同侧取代时,空间位阻较大,本文主要对NO3从异侧取代噻吩环(标记为R5)进行研究.NO3以SN2从异侧取代2-甲基噻吩时,经过渡态TS5向产物P5+CH3NO3的转化,伴随着C—C键的断裂和C—O键的生成.此过程需要越过的能垒为216.14kJ/mol,且为强吸热反应.因此,从热力学和动力学因素考虑异侧取代噻吩环均不易进行.
2.1.3 加成-消去反应 NO3与2-甲基噻吩发生加成-消去反应时主要存在两类反应机理,将噻吩环C—S键断裂的反应标记为R6、R7和R8,未断裂的反应标记为R9和R10.各通道的结构参数和能量信息如图2和表1所示.
NO3中一个O加成到2-甲基噻吩环上的C1时,首先生成复合物IM1A,释放出20.44kJ/mol的能量.再由复合物IM1A经过渡态TS6-1向中间体IM1转化,伴随着C—O键的生成.反应过程中C—O键长变化为2.378(IM1A)→2.083(TS6-1)→1.833(IM1),此过程仅需要越过5.46kJ/mol的能垒.由中间体IM1再经过渡态TS6-2向产物P6转化,同时伴随C—H键和NO3中的N—O键的断裂,此过程需要能垒为182.66kJ/mol,且是强吸热过程.从势能面上可以看出,该步骤为R6的速控步骤.显然,该通道不易进行.
NO3中一个O加成到2-甲基噻吩环上的C2(R7)和C3(R8)的反应与R6过程基本相似,不同之处是NO3中的O直接经过渡态TS7-1和TS8-1加成到噻吩环上,该过程所需的能垒均较小,然后经过渡态TS7-2和TS8-2向产物P7和P8转化,需要越过的能垒分别为139.20和144.58kJ/mol,显然,R7和R8均不易进行.
NO3中一个O与2-甲基噻吩环上的C1直接相连,经过无势垒过程首先形成复合物IM4,释放出47.26kJ/mol的能量.从复合物IM4出发,经过3种不同的反应机理,分别形成产物P10+NO2和P11+NO2,其中产物P10+NO2的形成经由两条反应路径,分别为IM4→TS9-1→P10+NO2(标记为R9-1)和IM4→TS9-2→P9→TS10→P10+NO2(标记为R9-2).P11+NO2的生成经由路径IM4→TS11→P11+NO2(标记为R10).
R9-1是由复合物IM4经过渡态TS9-1向产物P10的转化,伴随着C—S键和N—O键断裂.从IM4到过渡态 TS9-1,C—S键长由1.848增加到1.965,N-O键长由1.425增加到1.975.此过程需要越过54.58kJ/mol的能垒.R9-2与 R9-1不同之处在于,首先经过渡态TS9-2使C—S键断裂,然后经过渡态TS10使N—O键断裂,此过程需要的能垒分别为36.61和4.23kJ/mol.显然,经由两步过程比一步直接生成产物P10更易发生.
与R9-2、R9-1不同的是,R10涉及C1上的 H原子转移到C2上,形成稳定的五元环结构P11,并释放出NO2,此过程仅需要越过30.92kJ/mol的能垒,且从IM4出发,释放出164.23kJ/mol的能量.因此,从热力学和动力学因素考虑,通道R10较R9-1和R9-2易进行,故R10是加成-消去反应的主通道.
2.2 速率常数的计算
采用 Wigner校正[17-18]的过渡态理论计算标题反应中各通道在298K下速控步骤的反应速率常数kn.各反应通道的速率常数列于表2.
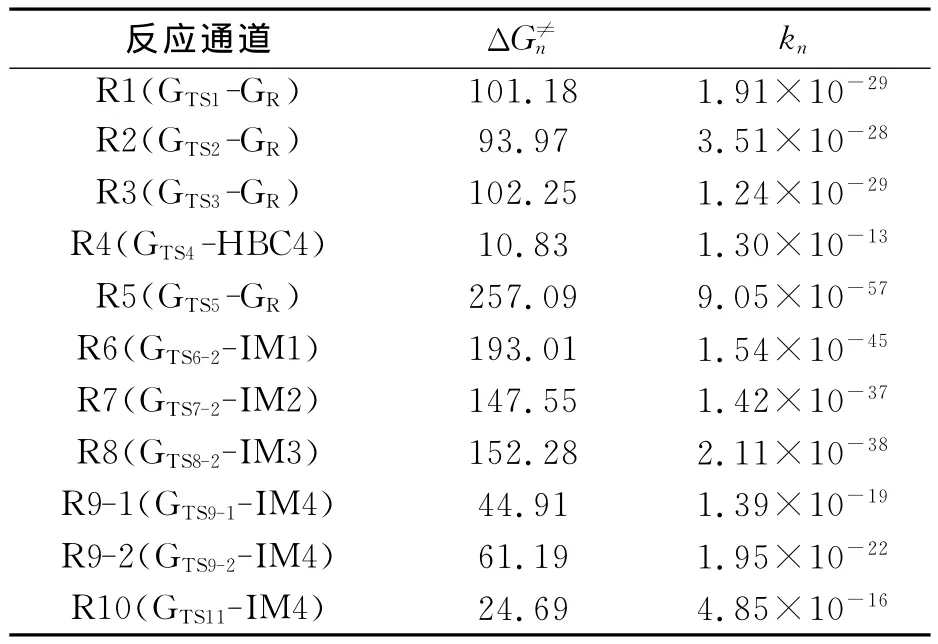
表2 298.15K下各反应通道速控步的吉布斯自由能(ΔG≠n)和速率常数(kn)Tab.2 Gibbs free energy(ΔG≠n,kJ/mol)and the rate constant(kn,cm3/molecule·s of the title reactions
从表2可以看出,在抽氢反应的4个通道中,k1~k4分别为 1.91×10-29、3.51×10-28、1.24×10-29、1.30×10-13cm3/(molecule·s),显然,R4的速率常数k4最大,其值与 Cabaas等人[4]用实验测得的速率常数(7.21×10-13cm3/(molecule·s))相近,因此R4为抽氢反应的主通道.在SN2反应中,速率常数极小,因此R5不易发生.在加成-消去反应中,从中间体IM4出发的三条路径 R9-1、R9-2、R10反应速率常数分别为1.39×10-19、1.95×10-22和4.85×10-16cm3/(molecule·s).通道 R10速率常数最大,因此加成-消去反应中,通道R10最有利进行.比较通道R4和R10的速率常数,k4≫k10,在动力学上,通道R4优于通道R10进行,故标题反应的主通道为R4.
3 结论
采用B3LYP/6-311++G(3df,2pd)//B3LYP/6-31+G(d,p)双水平方法构建反应体系的势能剖面图,通过分析反应机理,并计算各反应通道速率常数.可得出以下结论:2-甲基噻吩与NO3的反应存在抽氢、SN2取代和加成-消去3类机理,共10条反应通道,从动力学与热力学角度分析,抽氢反应中R4为标题反应的主通道,该通道的速率常数为1.03×10-13cm3/(molecule·s),与 Cabaas测定的实验值(7.21×10-13)接近.此外,甲基取代对噻吩与NO3的反应有显著影响,相对于甲基上的氢环上氢不易被抽取.这一结果为2-甲基噻吩与NO3反应的实验研究提供了有价值的参考信息.
[1]Lee J H,Tang I N.Absolute rate constants for the hydroxyl radical reactions with ethane,furan,and thiophene at room temperature[J].Journal of Chemical Physics,1982,77(9):4459-4463.
[2]Westerholm R N,Alm N J,Li H,et al.Chemical and biological characterization of particulate-,semivolatile-,and gas-phase-associated compounds in diluted heavyduty diesel exhausts:a comparison of three different semivolatile-phase samplers[J].Environmental Science and Technology,1991,25(2):332-338.
[3]Kirton P J,Ellis J,Crisp P T.The analysis of organic matter in coke oven emissions[J].Fuel,1991,70(12):1383-1389.
[6]Parthiban S,Lee T J.Theoretical study of XONO2(X=Br,OBr,O2Br):Implications for stratospheric bromine chemistry[J].Journal of Chemical Physics,2000,113(1):145-152.
[7]Lee T J,Mejia C N,Beran G J O,et al.Search for stratospheric bromine reservoir species:theoretical study of the photostability of mono-, tri-, and pentacoordinated bromine compounds[J].Journal of Physical Chemistry A,2005,109(36):8133-8139.
[8]Atklnson R,Aschmann S M,Wlner A M,et al.Rate constants for the gas-phase reactions of nitrate radicals with furan,thiophene,and pyrrole at 295 1Kand atmospheric pressure[J].Environmental Science and Technology,1985,19(1):87-90.
[9]Atkinson R,Aschmann S M,Pitts Jr J N.Rate constants for the gas-phase reactions of the nitrate radical with a series of organic compounds at 296 2K[J].Journal of Physical Chemistry,1988,92(12):3454-3457.
[12]Zhang Weichao,Wang Tao,Feng Changjun,et al.Computational studies on the mechanisms for the gasphase reaction between thiophene and NO3[J].Chemical Physics Letters,2008,467(1-3):52-57.
[13]Lee C,Yang W T,Parr R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Physical Review B,1988,37(2):785-789.
[14]Gonzalez C,Schlegel H B.An improved algorithm for reaction path following[J].Journal of Chemical Physics,1989,90(4):2154-2161.
[15]Gonzalez C,Schlegel H B.Reaction path following in mass-weighted internal coordinates[J].Journal of Physical Chemistry,1990,94(14):5523-5527.
[16]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian03[CP].Revision C.02,Gaussian,Inc.,Wallingford CT,2004.
[17]Resende S M,Orne llas F R.Atmospheric reaction between the HS radical and chlorine[J].Chemical Physics letters,2000,318(4/5):340-344.
[18]Du Benni,Zhang Weichao,Feng Changjun,et al.Thermodynamic and kinetic investigations on the reaction of O(3P)with HNO[J].Computational and Theoretical Chemistry,2004,712(1/3):101-107.
[19]Duncan W T,Bell R L,Truong T N.The Rate:program for ab initio direct dynamics calculations of thermal and vibrational-state-selected rate constants[J].Journal of Computational Chemistry,1998,19:1039-1052.
