逐级提取-高效液相色谱法快速测定植物组织中8 种有机酸
2014-12-26黄天志王世杰刘秀明吴沿友罗绪强
黄天志, 王世杰 , 刘秀明, 刘 虹, 吴沿友, 罗绪强
(1. 中国科学院地球化学研究所,环境地球化学国家重点实验室,贵州 贵阳550002;2. 中国科学院普定喀斯特生态系统观测研究站,贵州 安顺562100;3. 中国科学院大学,北京100049;4. 贵州师范学院,贵州 贵阳550018)
草酸是广泛存在于植物体中的简单二元羧酸,参与植物体内多种生理代谢过程,主要以水溶态和不溶于水的结晶态形式存在,其中以结晶态存在的草酸可以固定植物体内高达90% 的钙[1,2]。研究表明,植物体内草酸可能的来源包括乙醇酸、乙醛酸氧化、L-抗坏血酸裂解氧化、草酰乙酸裂解等途径,但参与草酸钙晶体形成的草酸具体来自于哪条途径却并不清楚[1,3,4]。因此,建立一种简单高效的方法测定乙醇酸、乙醛酸等草酸合成前体及不同形态草酸的含量,对研究草酸合成途经及其对应的生理功能具有重要的意义。
草酸等有机酸常用的分析方法有毛细管电泳法[5-7]、离子排斥色谱法[8]以及最常用的高效液相色谱法[9,10]。采用高效液相色谱法同时测定样品(食品、饮料、发酵产品及植物组织等)中的草酸及其他多种有机酸的方法常有报道,但同时测定草酸、乙醇酸及乙醛酸的方法还鲜有报道。目前有机酸的测定方法中,针对同一样品分形态提取并测定草酸等有机酸的研究还很少,仅侧重于水溶性有机酸的提取测定[11,12],或者用盐酸、硝酸等无机酸提取测定不同形态有机酸的总含量[3,13]。在需要不同形态草酸等有机酸的含量时,往往需要建立两种不同的色谱条件,分别测定酸溶态含量和水溶态含量,然后计算获得所需形态有机酸含量,这种方法在准确性和操作简便性方面都有待提高。此外,在同时测定草酸、酒石酸、乙醇酸、乙醛酸等的过程中,也存在一些需要解决的问题,例如以盐酸和硝酸等无机酸作为提取介质时,无机酸本身的吸收容易干扰测定的结果[13];草酸、酒石酸获得基线分离的难度较大[11];乙醇酸、乙醛酸需要先衍生后测定[10,14]等。本方法首先以水为提取介质,提取了水溶态草酸等8 种有机酸,而以草酸钙结晶等形态存在的草酸不溶于水,保留在固态残渣中;然后以稀盐酸为提取介质提取了酸溶性结晶态草酸。测定时采用磷酸二氢钾水溶液作为流动相,通过控制不同测定时段流动相的流速,在5 min 内完成了8 种有机酸的分离测定,并且在水溶态有机酸获得基线分离的同时,相同色谱条件下测定酸溶态草酸,较好地去除了盐酸对酸溶态草酸测定的干扰。
1 实验部分
1.1 仪器、试剂与材料
Agilent 1290 UPLC 液相色谱系统,配备二元泵(G4220B)、高性能自动进样器(G4226A)、柱温箱(G1316C)、二极管阵列检测器(G4212A);pHS-3C型精密酸度计(上海雷磁);超微量天平Cubis(德国Sartorius);0.22 μm 有机系滤膜(美国Whatman公司)。
8 种有机酸标准品均为色谱纯,草酸、乙酸、一水乙醛酸为Sigma-Aldrich 公司产品,乙醇酸、酒石酸、琥珀酸为Fluka 公司产品,苹果酸、柠檬酸为Supelco 公司产品;甲醇、乙腈为色谱纯,Dikma 公司出品;磷酸为色谱纯,磷酸二氢钾、盐酸、草酸钙为优级纯,均为天津科密欧公司产品;实验用水均为超纯水(18.2 MΩ·cm,Millipore)。
1.2 实验步骤
1.2.1 标准溶液制备
准确称取一水乙醛酸124.4 mg,草酸、乙醇酸、苹果酸、酒石酸、琥珀酸、柠檬酸各100 mg,用超纯水分别溶解并定容到50 mL;准确量取191 μL 乙酸,用超纯水定容到100 mL。由此获得2 g/L 的单一有机酸储备液,4 ℃下避光保存。使用时根据所需浓度用超纯水稀释,或混合配制混合酸。另外,准确量取盐酸4.106 mL,用超纯水定容到100 mL,配制成0.5 mol/L(18.23 g/L)的盐酸溶液备用。
1.2.2 样品前处理
所使用的植物样品为温室水培获得的诸葛菜(十字花科诸葛菜属,又名二月兰,学名Orychophragmus violaceus)的叶片和根。将叶片和根鲜样用超纯水冲洗干净,吸尽表面水分,各称取1.00 g。加入5 mL 超纯水研磨匀浆,将匀浆液全部转入离心管,30 ℃下振荡提取15 min,提取液于12 000 r/min 转速下离心10 min,收集上清液。加入5 mL超纯水冲洗离心管中残渣,同条件提取并离心收集上清液,该过程重复3 次。用超纯水将收集到的上清液定容到25 mL,此过程提取液为8 种水溶态有机酸样品(提取液Ⅰ)。向上述提取残渣中加入5 mL 0.5 mol/L HCl 溶液,80 ℃水浴振荡提取1 h,12 000 r/min 离心10 min 后收集上清液。采用5 mL 0.5 mol/L HCl 溶液冲洗残渣,同条件提取,重复3 次,用超纯水定容提取液至25 mL,此提取液为不溶于水的草酸样品(提取液Ⅱ)。分别取1.5 mL上述两种提取液经0.22 μm 滤膜过滤,测定提取液Ⅰ中8 种可溶性有机酸的含量,在相同色谱条件下测定提取液Ⅱ中酸溶态草酸的含量。
1.3 实验条件
Hypersil ODS (200 mm ×4.6 mm,5 μm)色谱柱,柱温30 ℃;KH2PO4水溶液(5 mmol/L,用H3PO4调pH 为2.8)作为流动相;进样量5 μL;检测波长210 nm。流速控制:0 ~1.4 min,1.4 mL/min;1.4 ~1.9 min,0.5 mL/min;1.9 ~5.0 min,1.4 mL/min。
2 结果与讨论
2.1 色谱条件的选择
2.1.1 流动相组成
硫酸[15,16]和磷酸盐缓冲体系[9-13,17-20]是有机酸分离常采用的流动相。因本实验使用HCl 作为提取介质,为避免其他无机酸的干扰,选择磷酸盐缓冲体系作为流动相,其中磷酸二氢钾因其较高的分离效率和极低的本底吸光度而被广泛采用。在采用KH2PO4溶液作为流动相分离有机酸时,其浓度从10 mmol/L 到36.7 mmol/L(0.5%)[9,11-13,17-19]均有报道,但用于分离草酸、乙醇酸和乙醛酸的KH2PO4溶液的浓度还少有报道。为寻找有效分离的最适KH2PO4溶液浓度,本实验配制了0、3、5、7、10、15、20、30、40 mmol/L 的KH2PO4溶 液(用H3PO4调 pH 为2.7)。色谱分离结果表明,KH2PO4溶液的浓度对苹果酸、琥珀酸、柠檬酸和乙酸的保留时间影响较小,在不同浓度的KH2PO4溶液条件下这4 种有机酸都能得到较好的分离。而草酸、盐酸、乙醛酸、酒石酸、乙醇酸保留时间接近,当KH2PO4溶液浓度大于10 mmol/L 时,草酸、盐酸、乙醛酸不能实现有效分离;当流动相中KH2PO4溶液浓度小于5 mmol/L 时,乙醇酸、酒石酸的分离效果受到盐酸干扰;KH2PO4溶液浓度介于5 mmol/L至10 mmol/L 之间时,8 种有机酸能较好地实现分离且不受盐酸干扰。从节约成本和环保的角度考虑,本实验最终选择5 mmol/L KH2PO4溶液作为流动相。
本实验还考察了在磷酸盐缓冲体系中加入有机相对分离效果和峰形的影响。结果表明,在5 mmol/L KH2PO4水溶液中加入甲醇会使草酸和HCl 的出峰时间延迟,当甲醇的体积比大于5% 时,草酸和乙醛酸不能有效分离,且甲醇的加入并没有显著地改善峰形,故本实验使用的流动相仅为KH2PO4溶液。
2.1.2 流动相pH 的选择
有机酸为弱酸,流动相pH 的大小直接影响其在磷酸盐缓冲体系中的解离程度。本实验测定以H3PO4调节流动相为不同pH 条件下的各有机酸保留时间(见图1)。结果发现,各有机酸保留时间对流动相pH 变化非常敏感,pH <2.6 时酒石酸、乙醇酸、乙醛酸分离效果不理想;pH 2.8 时各有机酸获得了最佳分离效果;pH >2.8 时乙醛酸、乙醇酸和酒石酸的分离度降低,且盐酸的干扰不能去除。因此选择pH 值为2.8 的KH2PO4溶液。

图1 流动相pH 对有机酸保留时间的影响Fig.1 Effect of pH value of mobile phase on the retention times of the organic acids
2.1.3 流速对分离效果的影响
流速直接影响各有机酸的保留时间。以5 mmol/L 的KH2PO4溶液(pH 2.8)为流动相,草酸、盐酸、乙醛酸、酒石酸都能有效分离但保留时间较为接近,需要控制较低流速来获得基线分离;而酒石酸、乙醇酸、苹果酸、琥珀酸、柠檬酸和乙酸峰形很好且保留时间间隔较大,因此需要调节不同的流速使所有组分获得基线分离的同时缩短分离时间。本实验采用分段控制流速的方法实现。0 ~1.4 min,流速1.4 mL/min,此时没有出峰;1.4 ~1.9 min,流速为0.5 mL/min,草酸、盐酸、乙醛酸和酒石酸实现较好的分离;1.9 ~5 min,流速为1.4 mL/min,其余5种有机酸得到快速且完全的分离。结果表明采用分段控制流速的方法,5 min 内能够完成8 种有机酸的有效分离,同时有效去除了盐酸对酸溶态草酸测定的干扰。有机酸标准样品的色谱图如图2 所示。
2.1.4 检测波长及柱温、柱压条件

图2 有机酸标准样品色谱图Fig.2 Chromatogram of organic acid standard mixtures
采用二极管阵列检测器扫描了各有机酸在190~400 nm 范围内的吸收,发现各有机酸在210 nm处有最大吸收,确定210 nm 为检测波长。实验测试了25、30、35 ℃3 个柱温,发现柱温对有机酸的分离没有显著影响,综合考虑设定柱温箱温度为30℃。实验过程中因为分时段控制流速,因此柱压随流速的变化而变化。在流速为1.4 mL/min 时,柱压范围为11 ~12 MPa;流速为0.5 mL/min 时,柱压范围为7 ~8 MPa。本实验所用UPLC 色谱系统柱压的高压限值为40 MPa,实验过程中柱压均保持在低于限值的合理范围内。
2.2 方法的线性范围和检出限
在1 ~2 000 mg/L 之间设置了10 个标准样品浓度梯度,重复进样3 次,以峰高Y(mAU)对质量浓度X(mg/L)进行线性回归得出各有机酸的线性方程及线性范围。选择线性范围下限浓度的有机酸标准样品重复进样7 次,并以信噪比为3(S/N =3)确定检出限,以线性范围下限浓度作为本实验实测样品的定量限。除乙醇酸、乙醛酸未能找到类似测定方法的检出限数据进行比较外,其他各目标有机酸在本实验所采用的色谱条件下的检出限均有不同程度降低,其中乙酸和琥珀酸的检出限水平略微高于文献[9,12]可查的最低值,但仍然处于较低水平。具体结果见表1。
2.3 回收率和精密度
回收率试验所用植物样品为供钙实验中供钙水平接近正常植物生境的一组样品。取相同根和叶片样品各3 份,其中一份作本底,测定各有机酸的含量和不同形态草酸的含量;另外两份分别添加两种水平的8 种有机酸标准溶液和1 mg 草酸钙(用以计算以稀盐酸为介质提取的酸溶态草酸的回收率)。按照1.2.2 节所述提取方法进行前处理,每份样品进行7 次平行测定。为了研究叶片和根可能存在的吸附效应的差异对回收率的影响,本实验分别计算了叶片和根的加标回收率和RSD,结果见表2。

表1 8 种有机酸的线性方程、相关系数、线性范围和检出限Table 1 Regression equations,correlation coefficients (R2),linear ranges and LODs of the eight organic acids

表2 植物叶片与根中有机酸的回收率及精密度(n =7)Table 2 Recoveries and precisions of organic acids from plant leaves and roots (n =7)
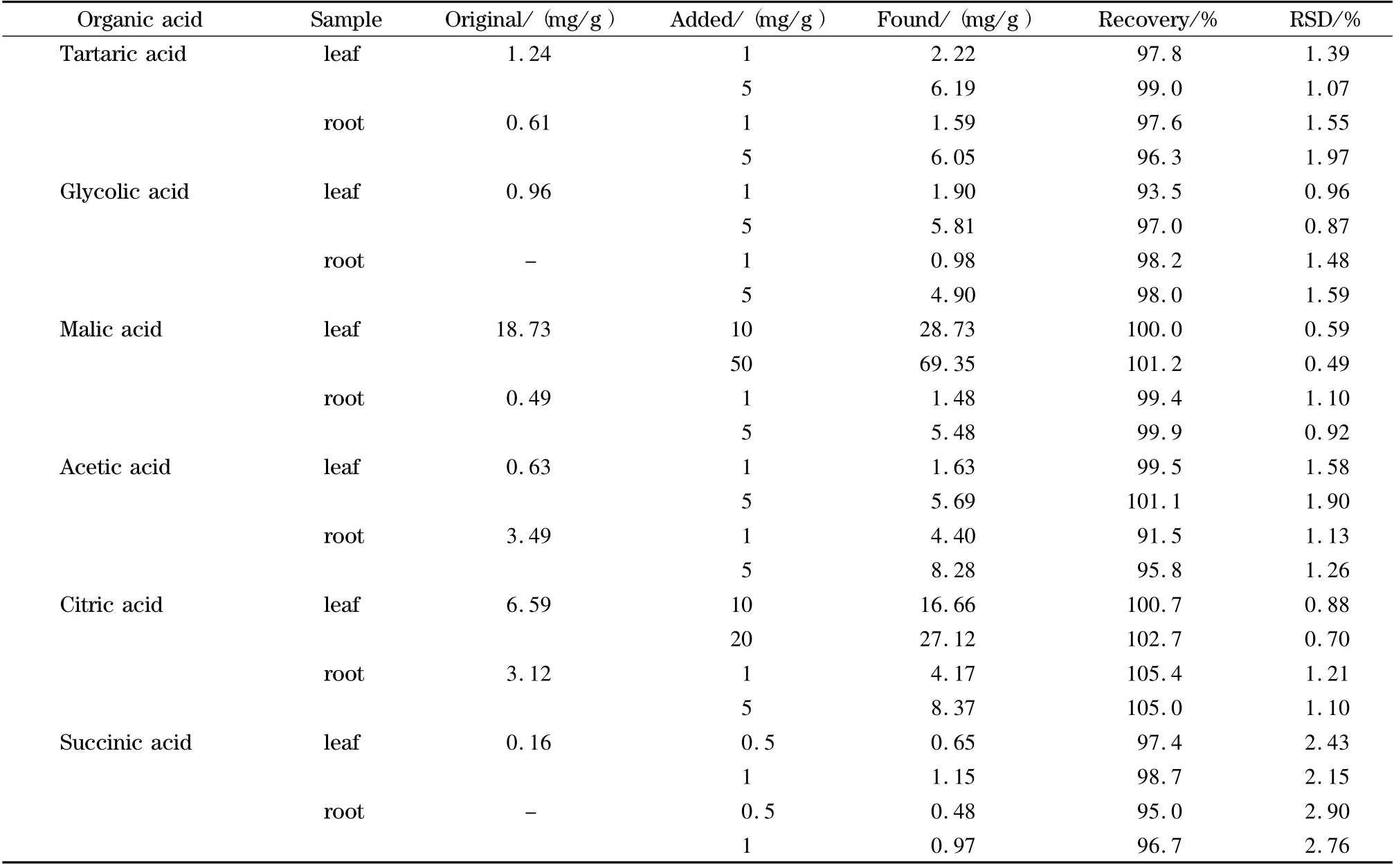
表2 (续)Table 2 (Continued)
2.4 实际样品测定
水培实验设计了梯度供钙水平,用以研究不同钙营养水平对诸葛菜有机酸组成和分布特征的影响。因为钙是植物生长的必需元素,所以本实验并没有设置钙的空白对照,而是设置了正常土壤供钙水平和极端高钙土壤的供钙水平,分别对应低供钙水平(5 mmol/L)和高供钙水平(50 mmol/L)。
采集不同供钙处理的样品,按1.2.2 节方法完成前处理,进样5 μL 测试分析,样品测定色谱图见图3,测定结果见表3。
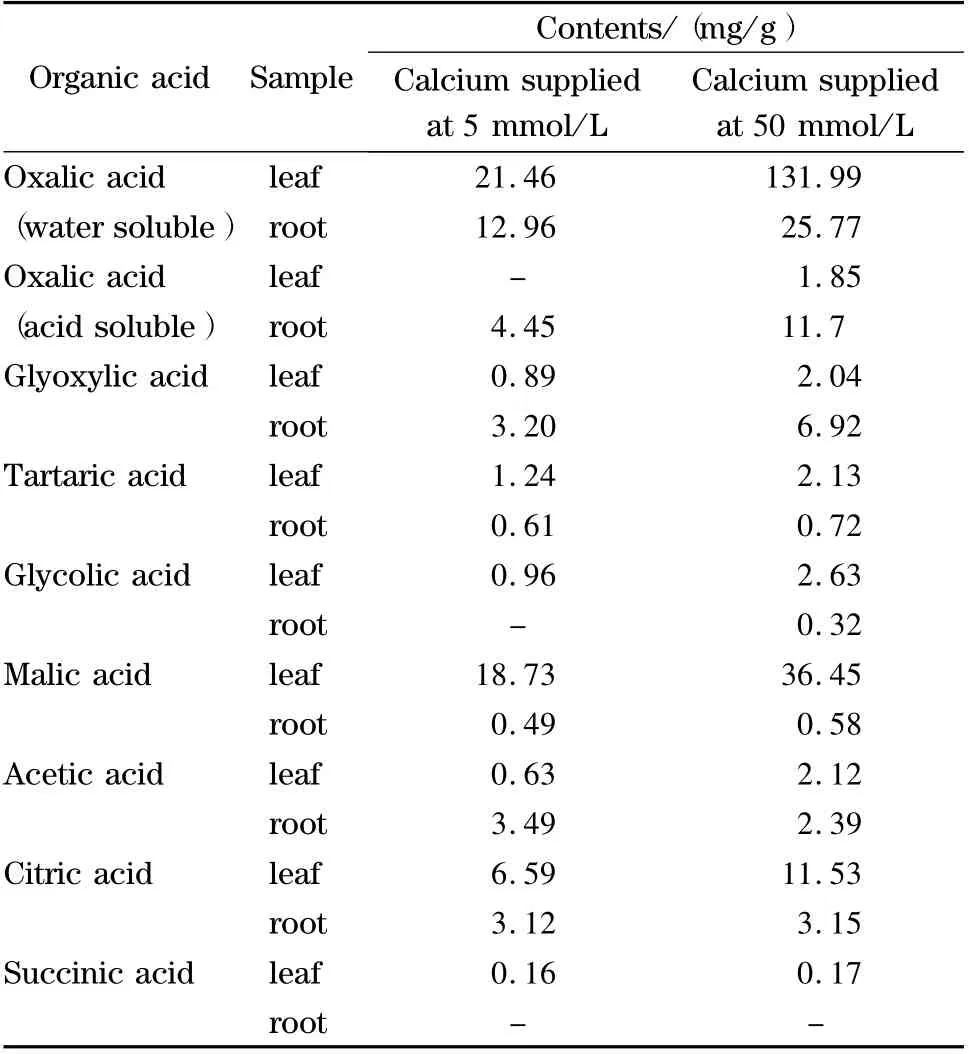
表3 实际样品中有机酸的含量Table 3 Contents of organic acids in real samples
结果表明:(1)叶片中水溶态有机酸含量与供钙水平有良好的响应关系;根组织中除乙醛酸和草酸外,其他水溶态有机酸含量对供钙水平不敏感;(2)不同组织中不同形态草酸的含量均与供钙水平具有良好的响应关系。
3 结论
本文建立了逐级提取、一步测定的方法,快速准确地提取测定了植物组织中不同形态的草酸以及和草酸合成相关的8 种有机酸。回收率试验和实际样品测定结果表明,本方法准确可靠,显著降低了目标有机酸测定的最低检出限,提高了方法的灵敏度,可以应用到草酸等有机酸的相关研究中。本方法避免了不同介质提取、不同色谱条件测定带来的误差,有效去除了无机酸对测定的干扰,5 min 内完成测定,提高了测定效率。同时,本方法测定酸溶态有机酸时,考虑到以无机酸作为提取介质所提取的成分较以水为提取介质提取的成分更为复杂,可能会干扰在本实验所采用的色谱条件下除草酸外的其他目标有机酸的测定,因此本方法只使用草酸数据,不使用其他有机酸的数据。在今后的工作中,随着测定仪器的不断升级,有机酸的测定方法仍然需要不断地改进和完善,需发展更加灵敏准确、方便快捷的分析测定方法。
[1] Franceschi V R,Nakata P A. Annu Rev Plant Biol,2005,56:41
[2] Chen Z,Geng H C,Wang S S,et al. Molecular Plant Breeding (陈峥,耿华春,王沙沙,等. 分子植物育种),2007,5(6(S)):105
[3] Xu H W,Ji X M,He Z H,et al. J Exp Bot,2006,57(9):1899
[4] Yu L,Jiang J Z,Zhang C,et al. J Exp Bot,2010,61(6):1625
[5] Tuma P,Samcova E,Stulik K. Anal Chim Acta,2011,685(1):84
[6] Han H F,Wang Q,Liu X,et al. Chinese Journal of Chromatography (韩海峰,王庆,刘霞,等. 色谱),2012,30(5):538
[7] Guan J,Wang H Z,Ren L Y,et al. Chinese Journal of Chromatography (关瑾,王慧泽,任丽艳,等. 色谱),2012,30(1):107
[8] Lin X J,Wei W,He Z G,et al. Chinese Journal of Chromatography (林晓婕,魏巍,何志刚,等. 色谱),2014,32(3):304
[9] Gao H Y,Liao X J,Wang S G,et al. Chinese Journal of Analytical Chemistry (高海燕,廖小军,王善广,等. 分析化学),2004,32(12):1645
[10] Ji X M,Yang C,Yang J,et al. Chinese Journal of Analytical Chemistry (籍秀梅,杨崇,杨军,等. 分析化学),2005,33(4):527
[11] Guo Y,Xiao Z P,Wang H,et al. Chinese Journal of Analytical Chemistry (郭瑛,肖朝萍,王红,等. 分析化学),2004,32(12):1624
[12] Jin G W,Zhang F F,Xue X Y,et al. Chinese Journal of Analytical Chemistry (金高娃,章飞芳,薛兴亚,等. 分析化学),2006,34(7):987
[13] Yu L,Peng X X,Yang C,et al. Chinese Journal of Analytical Chemistry (俞乐,彭新湘,杨崇,等. 分析化学),2002,30(9):1119
[14] Keates S E,Tarlyn N M,Loewus F A,et al. Phytochemistry,2000,53(4):433
[15] Ma R,Ouyang J,Li X,et al. Chinese Journal of Chromatography (马瑞,欧阳嘉,李鑫,等. 色谱),2012,30(1):62
[16] Guo Y,Liang J,Li M M,et al. Food Science(郭燕,梁俊,李敏敏,等. 食品科学),2012,33(2):227
[17] Lu M,Zhang S Y,Zhang W N,et at. Food Science (陆敏,张绍岩,张文娜,等. 食品科学),2012,33(14):235
[18] Cawthray G R. J Chromatogr A,2003,1011(1/2):233
[19] Zotou A,Loukou Z,Karava O. Chromatographia,2004,60:39
[20] Lin L C. Applied Chemical Industry (林灵超. 应用化工),2013,42(4):756
