基于部分酸水解-亲水作用色谱-质谱的黄芪多糖结构表征
2014-12-24辛华夏李芳冰梁鑫淼
梁 图, 傅 青, 辛华夏, 李芳冰, 金 郁, 梁鑫淼,2*
(1. 华东理工大学药学院,上海200237;2. 中国科学院大连化学物理研究所,辽宁 大连116023)
糖类化合物是能量的主要来源,除了提供能量,研究发现糖类化合物在人生命过程中发挥着至关重要的作用,对人体生理活动产生各种影响。因此,以糖类化合物为基础的糖类药物成了制药公司和生物技术公司长期关注的焦点。多糖是糖类化合物的一种,在过去的几十年中,植物性多糖特别是来自中药的水溶性多糖,由于其具有广谱治疗和低毒性特点,引起了广泛关注。从人参、三七、黄芪、冬虫夏草、当归、灵芝等中药中提取的多糖具有免疫调节、抗肿瘤、抗氧化、抗病毒、抗炎等生物活性,使之成为天然药物及保健品研发中的重要组成部分。虽然有不少的中药多糖已应用于临床,但由于其化学组成不明确,质量很难控制,造成药效重复性较差,不符合国际规范,因而也难以在医药领域占主要地位。为了使国际社会更加广泛地接受多糖药物在“自然疗法”方面的价值,需要发展简单而可靠的分析方法用于中药多糖的表征。然而,多糖结构复杂且相对分子质量巨大,其表征是一个大的挑战。
由于多糖的活性与多糖的相对分子质量、单糖组成、构型、糖苷键位置及三级结构相关,所以可使用色谱及光谱学方法检测中药多糖的结构特点,对其进行表征[1]。虽然获取结构信息是明确表征多糖的有效手段,但这个过程需要耗费大量精力,同时需要数量较多的多糖纯品,过程复杂、困难和耗时。在多糖分析中常使用蒽酮-硫酸法或苯酚-硫酸法等测定多糖含量,也可作为多糖表征的方法之一,多糖含量的变化可反映出不同产地、不同采收时期、不同栽培环境等方面的差异[2-4]。但该方法的选择性较差,不能有效鉴别出多糖制品中的一些掺假品[5];同时,该方法也无法直观反映出多糖中糖单元组成、个数和比例分布等方面的信息。因此有必要发展新的方法用于中药多糖的表征。
受蛋白质组学研究启发,可采用“自下而上”法完成对中药多糖的表征。先将中药多糖降解为特征性的寡糖片段,然后使用色谱以及色谱-质谱联用方法完成对特征性寡糖片段的分离和表征研究。酸水解是多糖降解中常用的方法之一,方法简单,易操作[6,7]。和酶解法相比,酸水解更加高效和快速[8]。由于酸水解过程一般与底物的结构无关,水解产物的聚合度仅与酸浓度和反应时间相关,因此具有普适性的特点,已被广泛用于多种多糖的降解,如壳聚糖[9]、木聚糖[10]和南瓜多糖[11]等。酸水解的目的是要获得充足数量的多糖降解碎片,因此要对部分酸水解的过程进行深入研究,考察酸水解的影响因素(包括酸浓度、水解温度和时间等),严格控制水解程度,保证重复、稳定地得到特征性的寡糖片段。
对于得到的多个特征性寡糖片段,需要采用合适的方法对其进行分离分析。而糖类化合物的分离和检测,也是糖学研究的难点问题之一。糖类化合物结构中包含大量的羟基,极性大,在传统的反相液相色谱固定相上很难保留,往往在死时间就被洗脱下来,难以得到有效分离。所以大多数的反相液相色谱法都需要使用衍生试剂对糖类化合物进行衍生,以增加其在反相色谱柱上的保留和提高检测灵敏度[12]。衍生步骤无疑增加了实验时间和方法的复杂程度。高效阴离子交换色谱-脉冲安培检测法适用于糖类化合物的直接分析[13]。但该方法使用高浓度、高pH 的非挥发性钠盐,对系统要求较高,并且不能直接与质谱联用。此外,研究发现亲水作用色谱(HILIC)适合用于糖类化合物的分离[14-16]。HILIC 使用极性分离材料为固定相,水溶性有机溶剂为流动相,克服了传统反相色谱对糖类化合物保留不足的弱点。同时,HILIC 方法使用高比例的有机溶剂和挥发性的缓冲盐,与质谱有好的兼容性,解决了离子交换色谱不能与质谱直接联用的问题。
本文选择中药黄芪为研究对象,发展基于部分酸水解-亲水作用色谱的黄芪多糖表征方法。首先对影响黄芪多糖部分酸水解的各种因素进行优化,建立黄芪多糖部分酸水解方法,得到特征性的寡糖片段;然后使用亲水作用色谱方法与质谱方法联用对黄芪特征寡糖片段进行分离和结构表征,获取黄芪多糖组成、连接等结构信息。
1 实验部分
1.1 仪器、试剂与材料
黄芪饮片购自上海雷允上药业有限公司,三氟乙酸(TFA,纯度≥99%)购自百灵威(中国)公司,无水乙醇、丙酮和石油醚购自国药化学试剂有限公司。实验用水来自Milli-Q 水纯化系统(Billerica,USA),乙腈(色谱纯)购自Fisher(Fair Lawn,USA),甲酸(色谱纯)购自Merck(Darmstadt,德国)。
色谱:Waters 高效液相色谱系统,包括Waters 2695 HPLC 泵、Waters 2420 蒸发光散射检测器(ELSD),数据记录使用Empower 工作站软件。
质谱:Thermo Scientific LCQ Fleet 质谱仪(Thermo,美国),配有电喷雾离子源(ESI),工作站为Xcalibur 2.1(Thermo,美国)。
1.2 多糖提取
如图1 所示,取20 g 黄芪饮片,粉碎后加水热浸提,滤液浓缩、醇沉、过滤,滤饼洗涤后得到黄芪多糖,干燥,备用。
1.3 多糖部分酸水解条件
称取10 mg 黄芪多糖,放入封管中,分别考察水解温度、时间及酸浓度的影响。
水解时间:三氟乙酸浓度为1.5 mol/L,水解温度为80 ℃,水解时间分别为2、3、4 和5 h,水解后氮气吹干,用1 mL 乙腈/水(50∶50,v/v)溶解,HILICELSD 分析。
酸浓度:水解温度为80 ℃,水解时间为4 h,三氟乙酸浓度分别为0.5、1.0、1.5 和2.0 mol/L,水解后氮气吹干,用1 mL 乙腈/水(50 ∶50,v/v)溶解,HILIC-ELSD 分析。
水解温度:三氟乙酸浓度为1.5 mol/L,水解时间为4 h,水解温度分别为40、60、80 和100 ℃,水解后氮气吹干,用1 mL 乙腈/水(50∶50,v/v)溶解,HILIC-ELSD 分析。
最终,黄芪多糖部分酸水解的适宜条件是水解温度80 ℃,三氟乙酸浓度1.5 mol/L,水解时间4 h。
1.4 分析条件
色谱柱:Click XIon (150 mm × 4.6 mm,5 μm,华谱新创科技有限公司)。
酸水解产物分析条件:流动相A 为H2O,B 为乙腈;洗脱梯度:0 ~40 min,85% B ~50% B;流速:1.0 mL/min。ELSD 检测参数:氮气压力2.07 ×105Pa,漂移管温度50 ℃,增益值100。
HILIC-MS 联用条件:流动相A 为H2O,B 为乙腈。洗脱梯度:0 ~40 min,85% B ~65% B;40 ~60 min,65% B。流速:1.0 mL/min,柱后分流比为1∶4。ESI 离子源接口,正离子模式一级全扫描,质量扫描范围为m/z 150 ~2 000。脱溶剂气为氮气,离子传输毛细管温度为300 ℃,鞘气流速为35 arb,辅助气流速为6 arb,喷雾电压为5.00 kV,毛细管电压为30 V。MSn 条件下分析时,毛细管传输能量为15~25 V。
2 结果与讨论
2.1 黄芪多糖部分酸水解方法的建立
使用“水提醇沉”法对黄芪饮片进行处理,得到黄芪多糖,具体处理步骤如图1 所示。为了得到特征性的寡糖片段,对影响水解过程的各种因素进行了研究,具体包括水解温度、时间和酸浓度。
首先考察水解时间对黄芪多糖降解程度的影响。水解后的多糖采用亲水作用色谱-蒸发光散射检测(HILIC-ELSD)方法对其进行分析,结果如图2所示。HILIC 模式下,黄芪寡糖在Click XIon 色谱柱上得到了较好的分离,按照极性由小到大的顺序依次被洗脱,最后的“大包峰”是未水解的黄芪多糖。当水解时间为2 ~3 h 时,黄芪多糖水解不够充分,大部分以多糖形式存在,寡糖含量较低;当水解时间增加至4 h 时,大部分的多糖开始水解,寡糖含量明显增加;水解时间进一步增加至5 h 时,谱图没有明显变化,由此推测,水解时间超过4 h 后再继续增加水解时间,对黄芪多糖的水解程度不再产生大的影响。所以4 h 为黄芪多糖水解的适宜时间。
接下来考察酸浓度对黄芪多糖降解程度的影响,结果如图3 所示。当三氟乙酸浓度为0.5 mol/L 时,水解不明显,大部分以多糖的形式存在;三氟乙酸浓度为1.0 mol/L 时,多糖水解速度加快,寡糖含量明显增加;当酸浓度增加至1.5 mol/L 时,大部分的黄芪多糖水解为寡糖;继续增加三氟乙酸浓度,谱图不再有明显变化。因此,确定黄芪多糖水解的适宜酸浓度为1.5 mol/L。
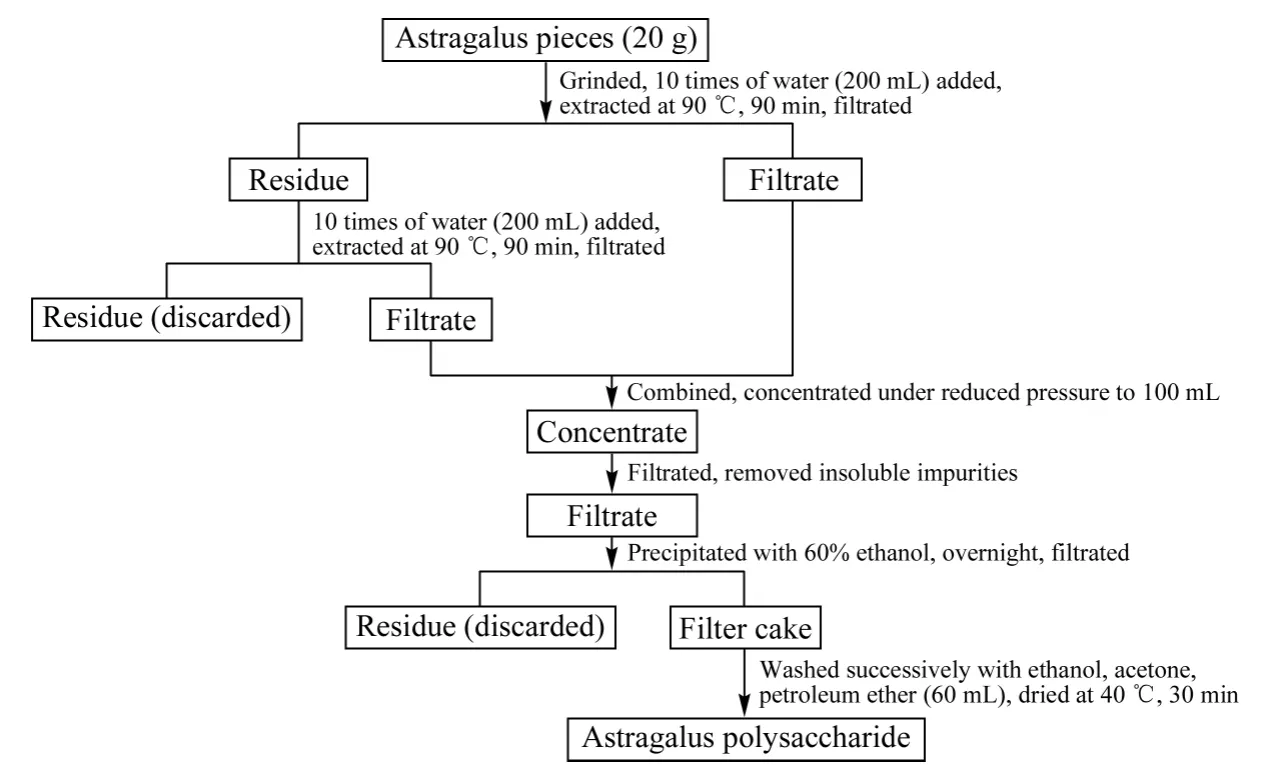
图1 黄芪多糖提取流程Fig.1 Process for polysaccharides preparation
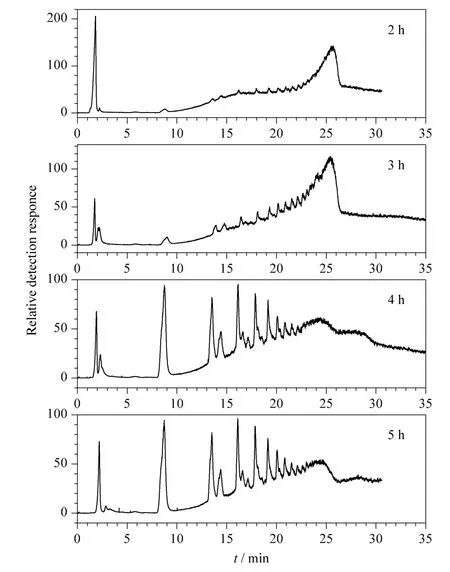
图2 水解时间对黄芪多糖部分酸水解的影响Fig.2 Effect of time on the partial acid hydrolysis of Astragalus polysaccharide
最后,考察水解温度对黄芪多糖降解程度的影响,结果如图4 所示。温度的变化对黄芪多糖水解程度的影响非常明显,40 ℃和60 ℃时,由于温度过低,黄芪多糖几乎不发生水解;当温度升高到80 ℃时,黄芪多糖迅速水解为寡糖,寡糖含量大幅度增加;当温度进一步升高时,黄芪多糖水解程度加剧,几乎全部水解为单糖和聚合度较小的寡糖。由此可见,多糖水解程度对温度的变化非常敏感,温度调节对控制多糖水解程度非常重要。所以,80 ℃为黄芪多糖水解的适宜温度,该温度下黄芪多糖水解程度适中,可产生大量特征性的寡糖片段。
2.2 黄芪多糖HILIC-MS 表征方法的建立

图3 TFA 浓度对黄芪多糖部分酸水解的影响Fig.3 Effect of TFA concentration on the partial acid hydrolysis of Astragalus polysaccharide

图4 温度对黄芪多糖部分酸水解的影响Fig.4 Effect of temperature on the partial acid hydrolysis of Astragalus polysaccharide

图5 黄芪多糖在正离子模式下的总离子流色谱图Fig.5 Total ion chromatogram of Astragalus polysaccharides in positive mode

表1 图5 中色谱峰1 ~8 的一级质谱数据Table 1 Data for the MS spectra of peaks 1 -8 in Fig.5
黄芪多糖经过酸水解后得到大量的寡糖片段,采用HILIC-MS 方法在正离子模式下对特征性寡糖片段进行分析,总离子流图如图5 所示。对图5 中色谱峰的质谱图进行分析发现,29 min 前为杂质峰,包括单糖、二糖以及黄芪提取物中的非糖杂质成分。如表1所示,对29 min 后的主要色谱峰1 ~8 做进一步分析。其一级质谱中糖类化合物在正离子模式下易产生[M +Na]+和[M +H]+离子,色谱峰1 ~8 对应的相对分子质量依次为666、828、990、1 152、1 314、1 476、1 638 和1 800。结合文献[17,18]报道判断,色谱峰1 ~8为聚合度(DP)4 ~11的中性葡寡糖。即本文提取得到的黄芪多糖主要为葡聚糖,被水解为DP 4 ~11 的葡寡糖。
对色谱峰1 ~8 做二级质谱表征,判断葡寡糖的连接方式。以[M +Na]+为母离子,进行二级质谱分析,结果如图6 所示。
色谱峰1 ~8 的二级谱图以[M +Na]+为母离子,母离子易丢失一分子的水,形成[M +Na -H2O]+碎片;丢失葡萄糖,形成一系列B 型碎片,m/z 1 643 (B10)、1 481 (B9)、1 319 (B8)、1 157(B7)、995(B6)、833(B5)、671(B4)、509(B3)和347(B2);丢失葡萄糖残基,形成一系列C 型碎片,m/z 1 661(C10)、1 499(C9)、1 337(C8)、1 175(C7)、1 013(C6)、851(C5)、689(C4)、527(C3)和365(C2)。完整的C 型和B 型碎片证明寡糖结构为线性。同时,[M +Na]+发生开环断裂,丢失60 Da,形成0.2Ai型碎片,m/z 1 763(0.2A11)、1 601(0.2A10)、1 439(0.2A9)、1 277 (0.2A8)、1 115 (0.2A7)、953(0.2A6)、791(0.2A5)、629(0.2A4)、467(0.2A3)和305(0.2A2),或者丢失120 Da,形成2.4Ai碎片,m/z 1 703(2.4A11)、1 541(2.4A10)、1 379(2.4A9)、1 217(2.4A8)、1 055(2.4A7)、893(2.4A6)、731(2.4A5)、569(2.4A4)、407(2.4A3)和245(2.4A2)。丢失60 和120 Da,为1→4连接的特征碎片。根据正离子模式下一级和二级质谱数据可知,本文中提取得到的黄芪多糖主要为1→4连接线性葡聚糖,该部分酸水解后产生DP 4 ~11 的葡寡糖。
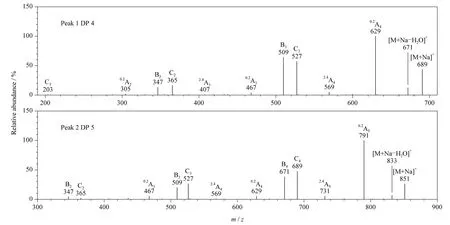
图6 图5 中色谱峰1 ~8 的二级质谱图Fig.6 Spectra of MS/MS fragmentation of peaks 1 -8 in Fig.5

图6 (续)Fig.6 (Continued)
3 结论
本文借鉴蛋白质组学“自下而上”法,完成对黄芪多糖的表征。结果表明,提取得到的黄芪多糖主要为1→4 连接线性葡聚糖,水解后得到DP 4 ~11的葡寡糖。该研究对其他中药多糖的表征具有一定的示范作用。进一步的工作将包括中药多糖水解产物中微量成分的分离和结构表征,以及中药多糖亲水指纹图谱的绘制,研究结果将有助于中药多糖的质量控制。
[1] Zhang B,Lin R C,Feng F. Chinese Pharmaceutical Affairs(张彬,林瑞超,冯芳. 中国药事),2004,18(9):566
[2] Deng G Q,Su Z N,Zhang J R,et al. Medical Information(邓桂球,苏振宁,张健润,等. 医学信息),2014,27(7):48
[3] Zeng L N,Yuan Y H,Huang S Y,et al. Fujian Forestry (曾丽娜,袁玉虹,黄淑燕,等. 福建林业),2014(2):25
[4] Xue M,Yang J Y,Dan S Y,et al. Journal of Mountain Agriculture and Biology (薛敏,杨继勇,但仕勇,等. 山地农业生物学报),2014,33(2):14
[5] Hu M H,Ma F L,Science and Technology Innovation Herald (胡明华,马方励. 科技创新导报),2010(30):10
[6] Reis A,Domingues M R M,Ferrer-Correia A J,et al. Carbohydr Polym,2003,53(1):101
[7] Daniel R,Chevolot L,Carrascal M,et al. Carbohydr Res,2007,342(6):826
[8] Akpinar O,Erdogan K,Bakir U,et al. LWT-Food Sci Technol,2010,43(1):119
[9] Yan X,Evenocheck H M. Carbohydr Polym,2012,87(2):1774
[10] Akpinar O,Erdogan K,Bostanci S. Carbohydr Res,2009,344(5):660
[11] Du B J,Song Y,Hu X S,et al. Int J Food Sci Technol,2011,46(5):982
[12] Wang H H,Dai J,Chen S W,et al. Food and Fermentation Industries (王浩豪,戴军,陈尚卫,等. 食品与发酵工业),2011,37(12):133
[13] Gangola M P,Jaiswal S,Khedikar Y P,et al. Food Chem,2014,154:127
[14] Alpert A J. J Chromatogr,1990,499:177
[15] Hemstrom P,Irgum K. J Sep Sci,2006,29(12):1784
[16] Liang X M. Chinese Journal of Chromatography (梁鑫淼.色谱),2011,29(3):191
[17] Ai L Z,Wu Y,Guo B H,et al. Shandong Food Fermentation (艾连中,吴艳,郭本恒,等. 山东食品发酵),2008(1):39
[18] He Y T,Pan X M. Food Science (何余堂,潘孝明. 食品科学),2010,31(17):493
