深圳大气中VOCs的二次有机气溶胶生成潜势
2014-12-14王扶潘刘芮伶黄晓锋何凌燕北京大学深圳研究生院环境与能源学院城市人居环境科学与技术重点实验室广东深圳518055
王扶潘,朱 乔,冯 凝,刘芮伶,黄晓锋,何凌燕 (北京大学深圳研究生院,环境与能源学院,城市人居环境科学与技术重点实验室,广东 深圳 518055)
深圳大气中VOCs的二次有机气溶胶生成潜势
王扶潘,朱 乔,冯 凝,刘芮伶,黄晓锋,何凌燕*(北京大学深圳研究生院,环境与能源学院,城市人居环境科学与技术重点实验室,广东 深圳 518055)
使用气溶胶生成系数法和产率法,对深圳四季5种芳香烃和异戊二烯所生成的二次有机气溶胶(SOA)量分季节计算并比较得出:四季中除春季外,其余三季产率法计算得到的 SOA 量均高于生成系数法计算的结果,四季所生成 SOA 的平均值分别为(2.48±2.02)μg/m3和(2.10±1.21)μg/m3. 2种方法计算结果均为夏季SOA的生成量最大,秋季和冬季次之,春季最小.生成系数法计算得出人为源和天然源的贡献分别为96%和4%,而产率法得到两种源的贡献分别为86%和14%.将生成系数法和产率法计算得到的夏季SOA值与实测值进行比较,发现计算值均低于实测值,分别占实测值的21%和31%.最后计算芳香烃和异戊二烯的自由基反应速率得出,与OH自由基反应是其生成SOA最主要的途径,比例为75%,与NO3自由基和O3的反应比例分别为22%和3%.在生成SOA速度上,苯乙烯的速度最快,苯的速度最慢.
气溶胶生成系数法;产率法;芳香烃;异戊二烯;二次有机气溶胶
大气中的挥发性有机物 (VOCs)如高碳烷烃、烯烃、芳香烃等由于其氧化态蒸汽压比还原态蒸汽压低,具有被氧化成二次有机气溶胶(颗粒物)SOA的潜力,可与大气中的 OH自由基、NO3自由基和O3等发生氧化反应而生成SOA[1].在北京、上海、珠江三角洲等地区的研究中均发现SOA是PM2.5的重要成分[2-4],而SOA中的苯环类(包括硝基苯、硝基酚、苯酚类)、脂肪族酸类、脂肪族硝酸酯类和羰基化合物类对人体健康和环境有重要影响[5],因此对SOA与前体物之间关系进行研究具有重要意义.
SOA的前体物包括挥发性有机物VOCs和半挥发性/中等挥发性有机物 S/IVOCs,而关于VOCs生成SOA的理论主要有气相氧化理论、气/固分配理论和颗粒相反应理论[6-9].研究者通过实测的VOCs浓度和烟雾箱实验来研究SOA的生成,在实验中将 VOCs引入烟雾箱并与其中的氧化剂反应,并加入一些颗粒物如(NH4)2SO4作为非均相成核表面,测量所生成的颗粒物,建立SOA 生成量与前体物之间的关系[10-13].Grosjean[5,14]假设VOCs生成SOA的产率是固定的,不随环境条件而变化,根据烟雾箱实验提出气溶胶生成系数FAC,来反映SOA与初始VOCs浓度间的关系,并对多种 VOCs的气溶胶生成系数进行了测定;Odum 等[15]对 FAC法进行了发展,考虑温度、颗粒物浓度等因素的影响,在烟雾箱实验中使用有机吸附分配的概念推测有机颗粒物的产率,对 VOCs的 SOA生成能力进行量化.国外的研究发现在计算得到的SOA中,芳香烃生成的SOA所占比例最大,如希腊雅典计算得到的SOA中 60%~90%的来自芳香烃的生成[16];美国南加州地区61%的SOA来自芳香烃氧化生成[13],在加拿大英属哥伦比亚地区,这个比例约为80%[17];异戊二烯来主要自于天然源的排放,且从全球尺度上来看是排放量最大的挥发性有机物,对 SOA有重要的贡献[18],因而本文主要研究芳香烃和异戊二烯的SOA生成潜势.
Jacobson等[19]的研究结果认为 VOCs中含碳数大于6的烷烃、烯烃、芳香烃和羰基化合物等才可以形成SOA,低碳数VOCs的氧化产物饱和蒸汽压太高而不能凝结成颗粒相,无法生成SOA,但Volkamer等[20]的研究表明乙二醛和乙炔等低碳的VOCs也具有生成SOA的能力.如前所述芳香烃和异戊二烯对SOA生成有重要的贡献,结合国内外关于生成系数和SOA生成产率的报道,以及实验室PTR-MS所监测的物种,本文所研究的VOCs物种包含有苯、甲苯、苯乙烯、C-8芳香烃、C-9芳香烃和异戊二烯.
本文根据 PTR-MS所测得的深圳四季VOCS的浓度,分别使用气溶胶生成系数法和气溶胶生成产率法对四季芳香烃和异戊二烯所生成的SOA进行计算比较分析,并将夏季计算得到的SOA值与实测值进行比较;最后计算芳香烃和异戊二烯的OH、NO3和O3自由基反应速率,探讨SOA的生成方式和速度.
1 采样与计算方法
1.1 采样
为了对深圳四季芳香烃和异戊二烯的 SOA生成潜势进行研究,作者在2011年8月10日至9月15日,2011年12月25日至2012年1月30日,2012年10月8日至11月11日和2013年3月1日至4月10日对VOCs浓度进行监测,分别代表深圳夏、冬、秋、春季的浓度水平,在每季采样中剔除异常值,均保证有30d的有效数据,采样地点位于北京大学深圳研究生院(22°35′28″N,113°58′30″E),校园及周边多植被,无明显人为源,该区域南面靠近留仙大道,东南面为南坪快速,附近以居民住宅区为主,兼有零星的工厂企业和商业设施.
在四季采样中均使用 PTR-MS—质子转移反应质谱[21](奥地利 Ionicon公司)在线对 VOCs的浓度进行测定,所监测的VOCs有19种,其中芳香烃物种有苯、甲苯、苯乙烯、C-8芳香烃和C-9芳香烃,以及异戊二烯.PTR-MS 基于VOCs与H3O+的质子转移反应,使用质谱检测器能快速测量生成的质子化VOCs离子(RH+),具有不需要前处理、时间分辨率高、灵敏度高等优点,可以在线测量质子亲和性高于水 VOCs物种,包括烯烃、醇、醛、酮、芳香烃和有机酸等.刘芮伶[22]在2011年夏季观测中对同一台PTR- MS与在线GC-FID/PID和DOAS所测结果进行了比对.发现三台仪器对甲苯的测量比对结果较好,变化趋势一致;苯、甲苯和 C8芳香烃的线性较好(r>0.95);GC-FID/PID的测量结果稍高于 PTR-MS测量结果,差异在30%之内.因而使用PTR-MS能很好的测定芳香烃物种的浓度.
1.2 SOA的计算
由于VOCs生成SOA的具体反应过程非常复杂,研究者在进行烟雾箱实验时,不考虑具体的反应过程,建立实测的VOCs浓度与SOA生成量之间的关系,使用参数化的方法计算不同条件下SOA的生成量[23-27].国内外研究中使用较多的方法有气溶胶生成系数法和气溶胶生成产率法.
1.2.1 气溶胶生成系数法 气溶胶生成系数FAC是SOA生成潜势的一种表达方式,对于可以生成SOA的VOCs组分i,所生成的SOA用式(1)进行计算:
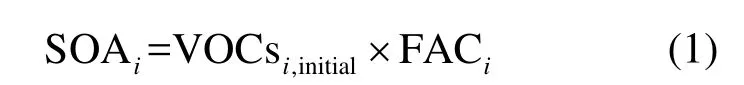
VOCs的初始浓度(VOCsi.initial)乘以相应的FAC系数就可以得到生成的SOA量.Grosjean在研究中认为VOCs生成SOA的生成系数FAC是固定的,本文使用的FAC值是Grosjean等[5,14]在大量烟雾箱实验数据和大气化学动力学数据的基础上提出的.对于VOCs初始浓度的计算,考虑到受体点测得的 VOCs往往是经过氧化后的VOCst,它与排放源排出的初始浓度之间的关系可通过式(2)来表示:
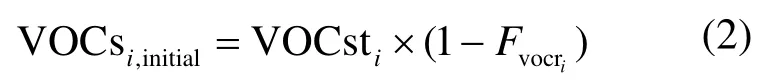
式中:VOCst是实测的浓度;FVOCr是VOCs物种参与反应的分数,%,该值使用Grosjean在烟雾箱实验中得到的数据.
对式(1)和式(2)进行整理综合,最终得到如下的计算式(3):
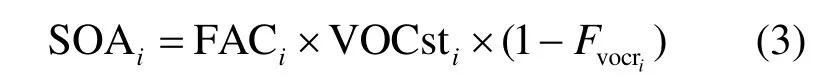
该方法的优点是计算简单,能从VOCs的排放清单或实测的环境浓度直接估算出 SOA的量,并可以反映各SOA前体物的贡献.根据Grosjean的假设:SOA 的生成只在白天(08:00~17:00)发生,且VOCs只与OH自由基发生反应,但实际VOCs生成SOA的途径还包括了与NO3自由基和O3的反应,因而根据FAC值结算的结果将低于实际值.
1.2.2 气溶胶生成产率法 Pankow[7-8]和Odum等[15]研究者认为 SOA 生成产率并非固定的,其值要受到温度、前体物、光照条件和 NOx浓度等因素的影响,结合气/固分配理论,最后发展成了SOA生成产率理论.VOCs在大气中的消耗是SOA生成的重要来源,因此以VOCs的消耗量作为基础,使用式(4)[26]对SOA的生成量进行计算:
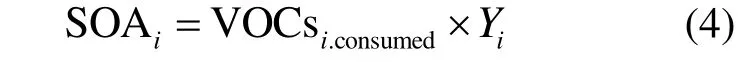
VOCs在大气中的消耗主要是与OH自由基的反应,de Gouw[28]提出了关于VOCs消耗量的计算式(5):
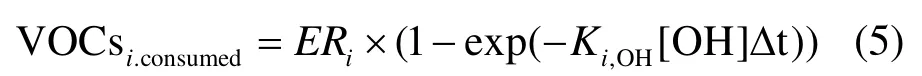
式中:VOCsi,consumed为 VOCsi的消耗量;ERi为VOCsi相对于CO的排放比;[OH]Δt为OH自由基暴露量;Yi为SOA产率[25].
Shilling 等[25]、Ng 等[26]、Carlton 等[27]对很多重要的 VOCs的 SOA生成产率进行了测定,发现在低 NOx状况下芳香烃的 SOA产率在0.3~0.4之间,高于在高 NOx状况下对应的产率,而且与颗粒物浓度没有关系,因而认为芳香烃氧化生成了挥发性很低的单一产物.本研究中使用的是 Ng[26]所测定的产率,且均为低 NOx浓度下VOCs的SOA产率,因而计算得到的SOA值是VOCs所能生成SOA的上限值,可以反映VOCs能生成SOA的最大潜势.
在计算光化学龄时倾向于选择2个 KOH值相差较大的 VOCs物种,如正丁烷/丙烷、甲苯/苯、二甲苯/乙苯、乙炔/CO,本研究中选择甲苯/苯来计算光化学龄[29-30].将OH自由基浓度[OH]与光化学龄Δt的乘积定义为 OH自由基的暴露量[31],使用式(6)进行计算:
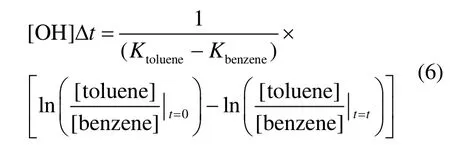
在式(5)计算 VOCsi的消耗量时需要知道各VOCsi相对于CO的排放比ERi、CO与OH自由基反应常数 KCO,二者可以利用式(7)进行多元非线性回归得到[28]:

式中:[VOCsi]是测到的 VOCsi浓度;[CO]为 CO浓度与CO背景浓度的差值;ERi为VOCsi相对于 CO的排放比;Ki、KCO分别为 VOCsi和 OH自由基与CO的反应常数;[OH]Δt为OH自由基暴露量.
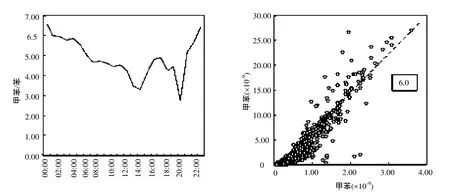
图1 春季甲苯与苯浓度比值的日变化和苯与甲苯的相关性(黑色虚线为二者的初始排放比)Fig.1 The diurnal variation of the ratio of toluene and benzene in the spring and their correlation. (The dotted line shows their initial emission ratio)
2 结果与讨论
2.1 芳香烃和异戊二烯的四季浓度水平
在春季观测期间6种VOCs的平均浓度为(7.67±5.34)×10-9夏季平均浓度为(11.13±8.59)×10-9,秋季平均浓度为(7.98±5.15)×10-9,冬季浓度为(8.30±8.78)×10-9,可以看出在 2012~2013 年深圳四季之中,6种VOCs物种浓度在夏季最高,冬季和秋季次之,春季最低,而四季浓度的平均值为(8.77±6.97)×10-9.
各VOCs物种的季节变化规律如图2所示:苯的浓度在春季和夏季相当,然后呈逐渐升高的趋势,冬季浓度最大,为(1.14±1.01)×10-9;甲苯是四季观测中浓度最高的物种且在夏季浓度最高,为(4.89±4.57)×10-9,秋季最低,为(3.46±3.19)×10-9.苯乙烯、C-9芳香烃和异戊二烯春季到夏季浓度增加,并在夏季达到四季浓度最大值,然后从夏季到冬季浓度逐渐降低;苯乙烯是浓度最低的VOCs,其最高浓度出现在夏季为(0.36±0.30)×10-9,其余三季相当;C-9芳香烃的最高浓度为(0.68±0.53)×10-9;异戊二烯主要来自于天然源的排放,因而在植物排放强烈的夏季出现浓度最大值为(0.75±0.59)×10-9;C-8芳香烃从春季到秋季浓度呈逐渐升高的趋势,然后降低并在冬季出现最低浓度,是排放量第二大的物种,其最高浓度出现在秋季,为(3.24±3.14)×10-9.
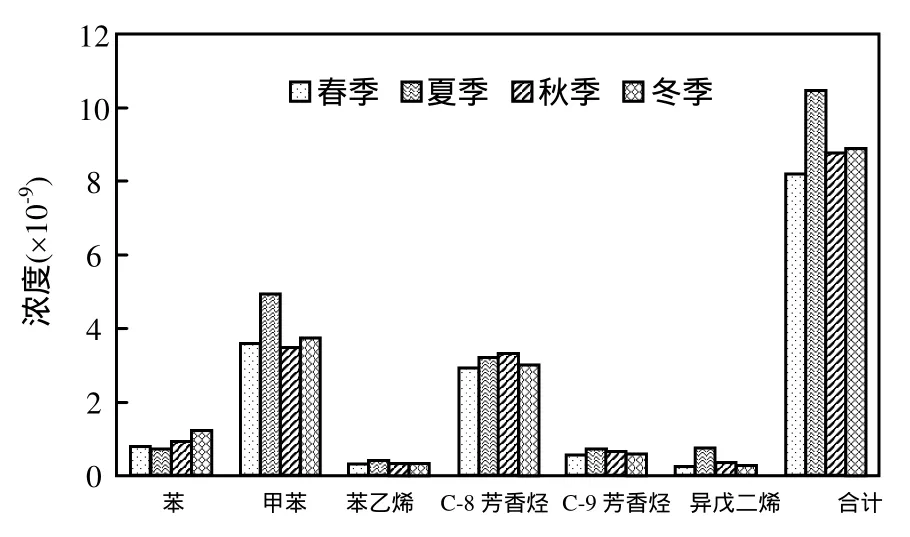
图2 深圳四季大气VOCs浓度变化Fig.2 Seasonal variations of atmospheric VOCs in Shenzhen
2.2 基于FAC法计算得到的SOA生成量
根据 Grosjean烟雾箱实验得到的气溶胶生成系数FAC和FVOCr值,结合式(3)计算得到深圳四季大气中各芳香烃和异戊二烯生成的SOA量,如表1所示.
从表1可以得出:四季中夏季生成的SOA量最大,为(2.70±1.55)μg/m3,冬季和秋季次之,分别为(1.92±1.26)μg/m3和(1.91±1.10)μg/m3,春季生成的SOA 量最少,为(1.85±0.93)μg/m3,全年 SOA 计算平均值为(2.10±1.21)μg/m3;5种芳香烃和异戊二烯所生成的 SOA与浓度呈正相关关系,即浓度越大则所生成SOA量也越大.甲苯由于浓度最高,SOA生成量最大,因而是对 SOA生成贡献最大的物种,其次是C-8芳香烃,浓度最低的苯,对SOA的生成贡献也最小;从全年看,6种物质对 SOA生成贡献的平均值分别为苯3%、甲苯46%、苯乙烯4%、C-8芳香烃33%、C-9芳香烃10%和异戊二烯4%;在春季和冬季异戊二烯对SOA生成贡献为3%,秋季为4%,夏季为7%,四季异戊二烯对SOA生成贡献的平均值为 4%.夏季异戊二烯的贡献最大,这与异戊二烯浓度夏季最高一致,可见天然源对 SOA的生成贡献受季节影响较大.以芳香烃为代表的人为源排放的VOCs对SOA生成贡献为96%,虽然没有对a蒎烯和β蒎烯生成的SOA进行计算,会低估天然源对 SOA的贡献,但仍然可以看出在深圳地区人为源的贡献远大于天然源的贡献.
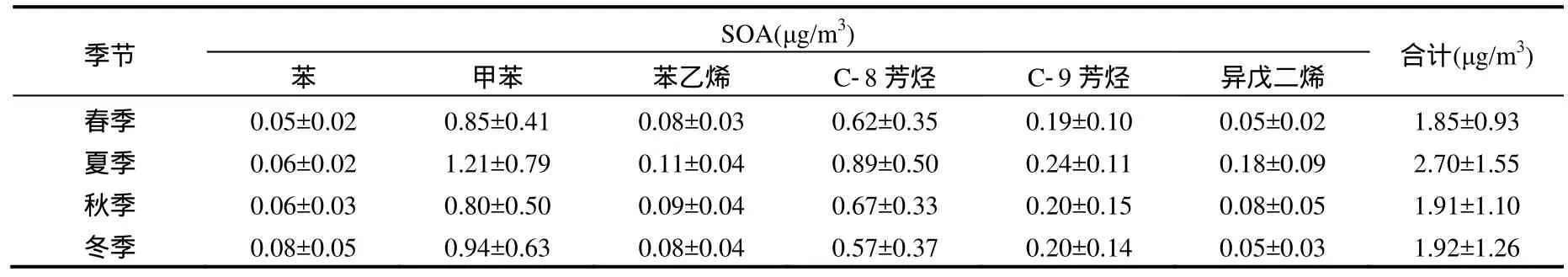
表1 FAC法计算得到的四季SOA生成量Table 1 The calculated SOA values of the four seasons by the FAC method
SOA的生成受到温度、湿度、有机气溶胶质量等很多因素的影响,因而用一个固定的 FAC值来表示SOA的产率会出现较大的偏差,但是通过FAC计算得到的SOA生成潜势仍能给出一些SOA的重要信息,比如SOA生成的大致数量级,以及各前体物的相对贡献等.
2.3 基于产率法计算得到的SOA生成量
使用Ng等[26]研究得到SOA生成产率,对深圳四季芳香烃和异戊二烯生成 SOA进行计算,结果如表2所示.
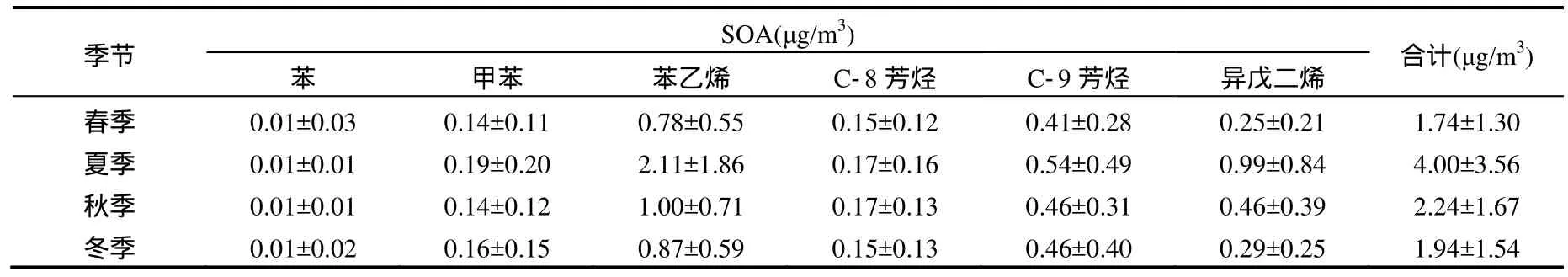
表2 产率法计算得到的四季SOA生成量Table 2 The calculated SOA values of the four seasons by the Yields method
根据产率法计算得到:四季中春季生成的SOA 最小,为(1.74±1.30)μg/m3,夏季生成的 SOA最高,为(4.00±3.56)μg/m3,秋季生成的 SOA 为(2.24±1.67)μg/m3,冬季生成 的 SOA 为 (1.94±1.54)μg/m3,虽然冬季 VOCs浓度高于秋季,但所生成的SOA较秋季低,全年SOA的计算平均值为(2.48±2.02)μg/m3;四季中,苯乙烯生成的 SOA值均为最大,是贡献最大的物种,而苯生成 SOA量最小,是贡献最小的物种;(3)从全年看,6种物质对SOA生成贡献的平均值分别为:苯小于1%、甲苯 6%、苯乙烯 48%、C-8芳香烃 7%、C-9芳香烃19%和异戊二烯14%;(4)春季天然源排放的异戊二烯对SOA生成的贡献为14%,夏季贡献最大且为20%,秋季贡献为17%,冬季贡献最小且为 5%,四季贡献的平均值为 14%,因而人为源排放的芳香烃对SOA生成平均贡献为86%,可以看出人为源的贡献远大于天然源的贡献.
在计算过程中发现,SOA计算值与浓度间并不存在一致的正相关,这是因为产率法的计算不仅要受到VOCs自身浓度的影响,还要受OH自由基暴露量、CO浓度的影响.影响因素的增多,会加大计算结果的不确定性,使得计算值和实测值间会存在较大的差距.
2.4 计算结果分析
对两种方法计算结果进行比较可以发现:四季中除春季外,其余三季产率法计算得到的SOA值均高于FAC法计算的结果,两种方法计算得到四季所生成SOA平均值分别为(2.10±1.21)μg/m3和(2.48±2.02)μg/m3,产率法计算值高于FAC法计算结果;两种方法计算结果均表明夏季芳香烃和异戊二烯生成的 SOA量最大,秋季和冬季次之,春季生成SOA量最小;产率法中四季异戊二烯对SOA生成贡献的平均值为 4%,产率法中异戊二烯贡献的平均值为 14%,因而人为源的贡献分别为96%和86%,两种方法均表现为人为源的贡献大于天然源的贡献;两者在评估物种对SOA生成贡献大小上存在差异,FAC法中对SOA生成贡献最大的物种是甲苯,而在产率法中贡献最大的为苯乙烯,贡献最小的均为苯;产率法对苯乙烯、C-9芳香烃和异戊二烯三种化学活性较强的VOCs生成的SOA计算值较FAC计算值高,因而产率法能更好地反映化学活性较强的 VOCs所能生成的SOA的水平.FAC法计算得到苯生成的SOA值约为产率法计算结果的5~8倍,可见产率法会低估活性较弱的VOCs生成SOA的能力.
在2011年夏季观测中,宫照恒[33]使用 AMS(气溶胶质谱仪)测得大气中 SOA的量为(12.98±10.06)μg/m3,则 FAC法计算值占实测值的 18%~30%,平均值为21%,产率计算值占实测值的15%~46%,平均为 31%,FAC法的计算过程较产率法简单,使用的是固定生成系数,误差也更大,产率法是对 FAC法的继承和发展,其计算结果更接近实测值.吕子峰[34]使用 FAC法计算得北京地区 VOCs对 SOA生成潜势的贡献 30%;Weitkamp[35]和Grieshop[36]通过烟雾箱实验结合产率法估算出的SOA生成量仅占实测值的2%~8%和5%~30%.在国内外的研究结中计算值与实测值的比在一个较大的区间波动,约为 0.5%~50%,由于计算过程中只考虑了与 OH自由基的反应,同时缺失其他VOCs生成SOA的数据,因而绝大多数的估算结果仅能解释不足一半的实测值.
不同的研究结果说明了参数化估算方法对SOA生成的量化有很大程度的不确定性,并且均表现为估算值比实测值低.本研究中产率法计算得到SOA值是芳香烃和异戊二烯生成SOA的上限值,但仍低于 AMS测得值,其主要原因是许多未测量的半挥发性有机物可能是SOA的重要前体物,并且很多VOCs的SOA生成系数和产率都还没有报道,使得计算值比实际值低.
2.5 自由基反应率对SOA生成的影响
在不考虑具体的反应过程的情况下,可以通过计算得到 VOCs所生成的 SOA,从反应机理上讲VOCs主要与OH自由基、NO3自由基和O3等发生反应而生成SOA,但3种自由基在SOA生成中的重要程度不尽相同,下面将使用深圳春季芳香烃和异戊二烯浓度数据计算自由基反应速率,来研究其生成SOA的主要方式和生成速度.VOCs的3种自由基反应速率可以用如下式进行计算:

式中:LOH,i、LNO3,i、LO3,i分别为 VOCsi的 OH、NO3、O3自由基反应速率;KOH,i、KNO3,i、KO3,i分别为 VOCsi与 OH、NO3和 O3自由基反应率常数[37];[VOCsi]为 VOCsi的浓度;[OH]、[NO3]和[O3][32]为 3种自由基浓度,在观测过程中没有对OH自由基的浓度进行测定,因而取文献中深圳地区OH自由基24h平均浓度2.5×106molecules/cm[22].

图3 不同VOCs的3种自由基反应速率比例Fig.3 The relative fractions of the reaction rates with the three radicals of different VOCs
从图3可以看出:芳香烃和异戊二烯与3种自由基反应中,OH自由基反应速率最大,NO3自由基反应速率次之,O3自由基的反应速率最小,说明大气中对其的去除主要是通过与 OH自由基反应,比例为 75%,NO3的去除作用占 22%,O3的去除作用占 3%;具体到每种物质,苯乙烯和异戊二烯由于有烯烃双键的存在,使得与NO3自由基和O3的反应消耗速率增加,其余芳香烃的OH自由基反应速率均大于 97%,NO3的贡献占极小的一部分,而基本不存在O3自由基消耗.
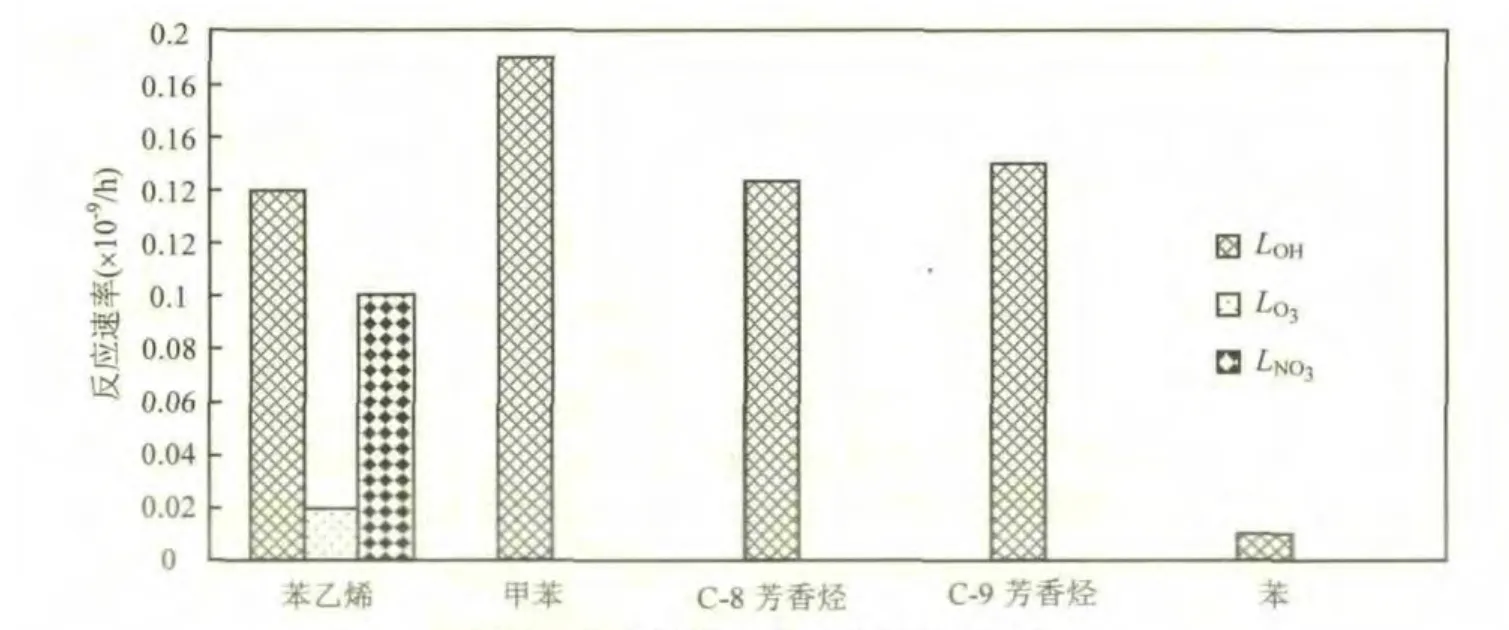
图4 芳香烃的3种自由基反应速率Fig.4 The reaction rates with the three radicals of different aromatics
如图 4所示,苯乙烯具有较高的 LOH值和LNO3值,同时也与 O3自由基发生反应,三自由基反应速率合计为 0.27×10-9/h,使得其在大气中的化学活性最为活跃,生成SOA的速度最快;甲苯、C-8芳香烃和C-9芳香烃的LOH值较高,分别为0.18×10-9/h、0.14×10-9/h 和 0.16×10-9/h,使得三者生成 SOA 速度较快,但是受其浓度的影响,所生成的SOA量与自由基反应速率不是正相关;苯的OH自由基反应消耗速率在3种自由基反应速率中最大,但也仅约 0.01×10-9/h,可见苯的大气化学反应活性很低,生成 SOA 的速度很慢,同时其浓度也最低,使得相同时间内所能生成的SOA量最低,对SOA生成贡献也最小.
综上,从反应方式上讲,芳香烃和异戊二烯与OH自由基反应是其生成二次有机气溶胶 SOA最主要的方式,如果是烯烃或含有烯烃双键,则与NO3自由基和O3的反应也是其生成SOA重要的途径;从生成速度上讲,苯乙烯生成 SOA 的速度最快,甲苯、C-8芳香烃和C-9芳香烃次之,苯生成速度最慢.
3 结论
3.1 四季除春季外,其余三季产率法计算得到的SOA值均高于FAC法计算的结果,两种方法计算得到四季所生成 SOA平均值分别为(2.10±1.21)μg/m3和(2.48±2.02)μg/m3,产率法计算值高于FAC法计算结果;夏季生成的SOA最大,秋季和冬季次之,春季最小;两种方法计算得到异戊二烯对SOA生成贡献的平均值分别为4%和14%,人为源的贡献分别为96%和86%,均表现为人为源的贡献大于天然源的贡献.
3.2 FAC法中对 SOA生成贡献最大的物种是甲苯,而在产率法中贡献最大的为苯乙烯,贡献最小的均为苯;产率法能更好的反映化学活性较强的VOCs所能生成的SOA的水平,但会低估活性较弱的VOCs物种生成SOA的能力.
3.3 FAC法计算值占实测值的21%,而产率计算值占实测值的 31%.产率法使用低 NOx浓度下SOA产率计算得到的SOA值是VOCs所能生成SOA 的上限,但仍较实际值低,说明使用的参数化方法估算SOA生成潜势有很大程度的不确定性.
3.4 芳香烃和异戊二烯主要通过与OH自由基反应而生成SOA,但苯乙烯和异戊二烯生成SOA的途径还包括了与O3和NO3自由基反应;在生成SOA速度上,苯乙烯的速度最快,甲苯、C-8芳香烃和C-9芳香烃次之,苯的速度最慢.
[1]唐孝炎,张远航,邵 敏.大气环境化学. [M]. 2版.北京:高等教育出版社, 2006
[2]Feng Y, Chen Y, Guo H, et al. Characteristics of organic and elemental carbon in PM2.5samples in Shanghai, China [J].Atmospheric Research, 2009,92(4):434-442.
[3]Lin P, Hu M, Deng Z, et al. Seasonal and diurnal variations of organic carbon in PM2.5 in Beijing and the estimation of secondary organic carbon [J]. Journal of Geophysical Research,2009,114(D2):D00G11.
[4]Hu W W, Hu M, Deng Z Q, et al. The characteristics and origins of carbonaceous aerosol at a rural site of PRD in summer of 2006[J]. Atmospheric Chemistry and Physics, 2012,12(4):1811-1822.
[5]Grosjean D. In situ organic aerosol formation during a smog episode estimated production and chemical functionality [J].Atmospheric Environment, 1992,26A:953-963.
[6]Kroll J H, Seinfeld J H. Chemistry of secondary organic aerosol:Formation and evolution of low-volatility organics in the atmosphere [J].Atmospheric Environment, 2008,42(16):3593-3624.
[7]Pankow J F. An absorption-model of gas-particle partitioning of organic-compounds in the atmosphere [J]. Atmospheric Environment, 1994,28(2):185-188.
[8]Pankow J F. An absorption-model of the gas aerosol partitioning involved in the formation of secondary organic aerosol [J].Atmospheric Environment, 1994,28(2):189-193.
[9]陈文泰,邵 敏,袁 斌,等.大气中挥发性有机物(VOCs)对二次有机气溶胶(SOA)生成贡献的参数化估算 [J]. 环境科学学报, 2013,33(1): 163-172.
[10]Alfarra M R, Paulsen D, Gysel M, et al. A mass spectrometric study of secondary organic aerosols formed from the photooxidation of anthropogenic and biogenic precursors in a reaction chamber [J]. Atmospheric Chemistry and Physics, 2006,6(12):5279-5293.
[11]Hao L Q, Wang Z Y, Huang M Q, et al. Effects of seed aerosols on the growth of secondary organic aerosols from the photooxidation of toluene [J]. Journal of Environmental Sciences,2007,19(6):704-708.
[12]Robinson A L, Donahue N M, Shrivastava M K, et al. Rethinking organic aerosols: semivolatile emissions and photochemical Aging [J]. Science, 2007,315(5816):1259-1262.
[13]George I J, Abbatt J P D. Chemical evolution of secondary organic aerosol from OH-initiated heterogeneous oxidation [J].Atmospheric Chemistry and Physic, 2010,10(12):5551-5563.
[14]Grosjean D, Seinfeld JH. Parameterization of the formation potential of secondary organic aerosols [J]. Atmospheric Environment, 1989,23:1733-1747.
[15]Odum J R, Jungkamp T P W, Griffin R J ,et al.The atmospheric aerosol-forming potential of whole gasoline vapor [J]. Science,1997,276(5309):96-99.
[16]Kourtidis K, Ziomas I. Estimation of secondary organic aerosol( SOA)production from traffic emissions in the city of Athens [J]. Global Nest, 1999,1(1):33-39.
[17]Barthelmie R J, Pryor S C. Secondary organic aerosols: formation potential and ambient data [J]. Sci. Total.Environ, 1997,205(2)-3:167-178.
[18]Claeys M, Graham B, Vas G, et al. Formation of secondary organic aerosols through photo oxidation of isoprene [J]. Science,2004,303:1173–1176.
[19]Jacobson M C, Hansson H C, Noone K J, et al. Organic atmospheric aerosols: review and state of the science [J]. Rev Eophys, 2000,38(2):267-294.
[20]Volkamer R, Martini F S, Molina L T D, et al. A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol [J]. GeophysicalResearchLetters, 2007,34(L19807),doi:1029/2007GL030752.
[21]Hansel A, Jordan A. Proton-transfer reaction massspectrometry—online trace gas-analysis at the ppb level [J].International Journal of Mass Spectrometry and Ion Processes 1995,149:609-619.
[22]刘芮伶.基于质子转移反应谱的大气含氧挥发性有机物气在线测量和来源研究 [D]. 北京:北京大学硕士学位论文, 2012.
[23]DeCarlo P F, Kimmel J R, Trimborn A, et al. Field-deployable,high-resolution, time-of-flight aerosol mass spectrometer [J].Anal Chem, 2006,78(24):8281-8289
[24]Kimmel J R, Farmer D K, Cubison M J, et al. Real-time aerosol mass spectrometry with millisecond resolution [J]. Int J Mass Spectrom, 2011, 303(1):15–26.
[25]Shilling J E, Chen Q, King S M, et al. Particle mass yield in secondary organic aerosol formed by the dark ozonolysis of alpha-pinene [J]. Atmospheric Chemistry and Physics, 2008,8(7):2073-2088.
[26]Ng N L, Kroll A. Chan W, et al.Secondary organic aerosol formation from m-xylene, toluene, and benzene [I], Atmospheric Chemistry and Physics, 2007,7(14):3909-3922.
[27]Carlton A G, Wiedinmyer C, Kroll J H. A review of Secondary Organic Aerosol(SOA)formation from isoprene [J]. Atmospheric Chemistry and Physics, 1999,9(14),4987-5005.
[28]de Gouw J A. Budget of organic carbon in a polluted atmosphere:Results from the New England Air Quality Study in 2002 [J],Journal of Geophysical Research Atmospheres,2005,110(D16305),doi:10.1029/2004JD005623.
[29]McKeenS A, Liu S C. Hydrocarbon ratios an photochemical of air mass [J]. Geophysical Research Letters, 1993,20(21):2363-2366.
[30]Grosjean E, Rasmussen R A. Toxic air contaminants in Porto Alegre, Brazil [J]. Environmental Science and Technology,1999,33(12): 1970-1978.
[31]Jimenez J L. Evolution of organic aerosols in the atmosphere [J],science, 2009,326(5959),1525-1529.
[32]袁 斌.挥发性有机物(VOCs)化学转化的量化表征及其应用研究 [b]. 北京:北京大学博士学位论文.
[33]宫照恒.基于高分辨质谱在线观测的2011深圳大运会前后PM1化学组成与粒径分布 [J]. 中国科学:化学, 2013,43(3):363-372.
[34]吕子峰,郝吉明,段菁春,等.北京市夏季二次有机气溶胶生成潜势的估算 [J]. 环境科学, 2009,30(4):969-975.
[35]Weitkamp E A, Sage A M, Pierce J R, et al. Organic aerosol formation from photochemical oxidation of diesel exhaust in a smog chamber [J]. Environmental Science and Technology, 2007,41(20):6969-6975.
[36]Grieshop A P, Logue J M, Donahue N M, et al. Laboratory Investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution [J]. Atmospheric Chemistry and Physics, 2009,9(4):1263-1277.
[37]Atkinson R, Baulch D L, Cox R A, et al. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II; gas phase reactions of organic species [J]. Atmospheric Chemistry and Physics, 2006,6:3625-4055.
The generation potential of secondary organic aerosol of atmospheric VOCs in Shenzhen.
WANG Fu-pan, ZHU Qiao,FENG Ning, LIU Rui-ling, HUANG Xiao-feng, HE Ling-yan*(Key Laboratory for Urban Habitat Environmental Science and Technology, School of Environment and Energy, Shenzhen Graduate School, Peking University, Shenzhen 518055,China). China Environmental Science, 2014,34(10):2449~2457
Both the fractional aerosol coefficients (FAC) and yields (Y) methods was wsed to calculate the secondary organic aerosol (SOA)production from atmospheric volatile aromatics and isoprene in ShenZhen. Throughant the year results from the Yields method were all larger than the results by the FAC method except in spring, and the total average SOA production values were (2.48±2.02)μg/m3and (2.10±1.21)μg/m3. The calculated SOA value in the summer was the largest, followed by in the autumn, winter, and spring. By the FAC method, the contribution from anthropogenic sources was 96%, while the contribution from natural sources was 4%. By the Yields method, contributions of anthropogenic and natural sources were 86% and 14%, respectively. During summer time, the calculated SOA values by the FAC and Yields methods only accounted for 21% and 31% of the measured SOA value. The OH radical reaction activities of aromatic hydrocarbons and isoprene were also calculated, and the results showed that the aromatic hydrocarbons' reaction with OH radicals was the main source of SOA with a percentage of 75%, while the percentages of NO3and O3were 22% and 3%.In terms of generating speed of SOA, styrene was found to have the fastest speed, while the speed of benzene was the slowest.
FAC method;Yields method;aromatics;isoprene;secondary organic aerosol
X513
A
1000-6923(2014)10-2449-09
2013-11-27
国家自然科学基金(U1301234;21177001);国家“973”项目(2013CB228503);深圳市科技计划
* 责任作者, 教授, hely@pku.edu.cn
王扶潘(1987-),男,四川绵阳人,北京大学环境与能源学院硕士研究生,主要从事大气挥发性有机物研究.
