NOTCH3基因剪切突变导致的CADASIL一家系报告
2014-11-23赵丹华贺大权杨旭金迪刘潇王朝霞袁云
赵丹华 贺大权 杨旭 金迪 刘潇 王朝霞 袁云
常染色体显性遗传性脑动脉病伴皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)是一种较常见的遗传性脑小血管病[1],于1993年由法国人 Tournier-Lasserve等首次提出[2]。该病多在中年发病,临床特征为反复发作的缺血性卒中、偏头痛、精神异常和皮质下痴呆[1]。头颅MRI显示多发的腔隙性脑梗死及广泛的白质疏松,尤其以颞极白质异常信号最有提示意义[1,3]。其特征性的病理改变为小动脉中层平滑肌细胞表面出现颗粒性嗜锇物质(GOM)沉积,是诊断本病的金标准之一[1]。
CADASIL由位于染色体 19p13.2-13.1 的NOTCH3基因突变所引起[4]。致病性NOTCH3基因突变也是确诊该病的金标准。根据HGMD数据库 (www.hgmd.cf.ac.uk/ac/index.php)显示,目前全世界已报道240余种突变,其中90%以上为错义或无义突变,还有少数为插入以及缺失突变,而剪切突变罕见,至今仅有3例[5-7]。我国目前已报道40余个CADASIL家系[8-12],全部为错义或缺失突变引起,尚无剪切突变的报道。本文通过分析1例CADASIL患者及其家系成员的临床、影像学特点,并对患者及部分家系成员进行NOTCH3基因检测,发现其存在NOTCH3基因剪切位点突变。
1 对象和方法
1.1 研究对象 收集1个CADASIL家系共5例患者的临床及影像学资料,家系系谱见图1。先证者经超微病理检查发现微小动脉的平滑肌细胞表面以及毛细血管周细胞表面存在GOM沉积,符合CADASIL诊断标准。

图1 CADASIL患者家系系谱图
1.2 方法
1.2.1 临床资料收集:回顾分析该家系先证者(Ⅲ-5)及家系成员中有症状患者的临床表现,主要包括发病年龄、首发症状及主要症状等;回顾分析先证者2次不同时期的头MRI表现。
1.2.2 NOTCH3基因突变检测:提取先证者及其兄妹(Ⅲ-6及Ⅲ-7)的外周血白细胞DNA。使用Primer 3软件对NOTCH3基因的热点突变区域2-12号外显子及其侧翼序列设计引物,具体引物序列及反应条件见参考文献[13]。聚合酶链反应(PCR)扩增目的片段,产物纯化后,用 ABI 3730XL自动测序仪进行直接测序。所有PCR产物均进行双向测序。同时对100名健康对照者进行NOTCH3基因4号外显子及其侧翼序列测序。
1.2.3 突变的反转录分析:用Trizol试剂一步法从患者左肱二头肌组织中提取总RNA,在 MMLV反转录酶作用下反转录得到第一链cDNA。以此cDNA为模板进行PCR反应,使用Primer 3设计引物覆盖NOTCH3基因2-4号外显子,上游引 物 序 列 为:5′-GTGTGCAAATGGAGGTCGTT-3′,下游引物序列为:5′-AAGGAGCCAGGTGTGTTGAG-3′。应用琼脂糖凝胶检测PCR产物并进行双向测序。
2 结果
2.1 临床及影像学表现 该家系中3代共出现5例患者,且每代均有发病者。先证者为46岁男性,40岁出现短暂性脑缺血发作;43岁患脑梗死,当时查头MRI示左侧脑桥点状新鲜梗死灶,同时双侧侧脑室旁白质、左侧胼胝体及双侧基底节可见多发陈旧腔梗灶,双侧侧脑室旁白质及额顶颞叶皮层下白质多发小片状长T2信号,双侧颞极白质及右侧外囊呈长T2信号(图2);46岁开始出现记忆力减退,复查头MRI示双侧基底节及半卵圆中心腔隙性脑梗死灶较以前增多,双侧侧脑室旁白质及额顶颞叶皮层下白质病变范围较前扩大,部分融合成片。先证者的弟弟(Ⅲ-7)30多岁时患“脑梗死”,目前遗留言语不清、记忆力减退、思维混乱表现;父亲(Ⅱ-3)约50岁时出现反应迟钝、记忆力减退及运动迟缓等,后因瘫痪卧床去世;姑姑(Ⅱ-2)曾患“脑梗死”,现已去世;祖父(Ⅰ-1)早年死亡,原因不详,去世前有动作迟缓及言语不清表现。所有患者均无高血压、糖尿病及高脂血症等疾病。
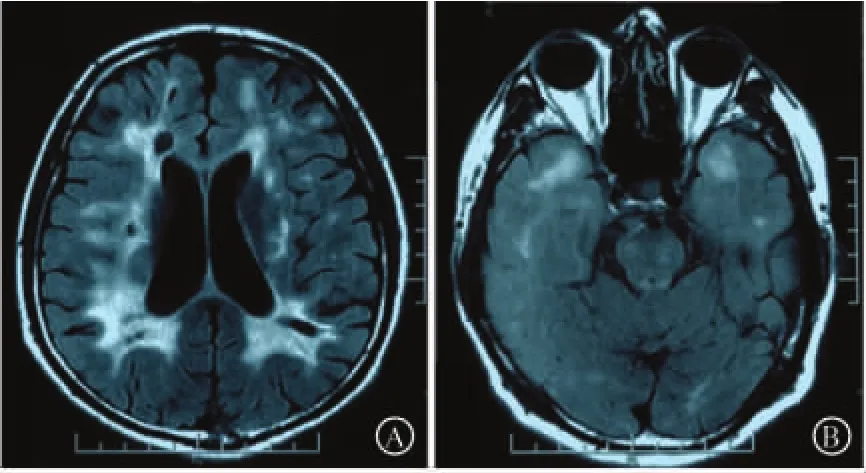
图2 先证者(43岁)患脑梗死时的头MRI表现:双侧侧脑室周围出现多发腔梗灶,同时可见脑白质变性(A);双侧颞极可见T2高信号(B)
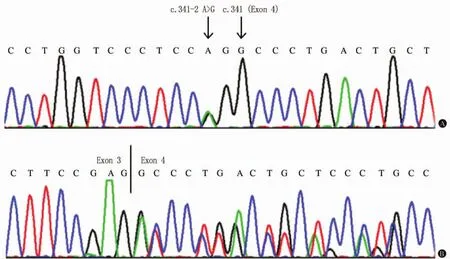
图3 先证者的NOTCH3基因测序分析
2.2 基因检测 先证者及其弟弟(Ⅲ-7)的NOTCH3基因3号内含子的第77位碱基存在A>G杂合突变(c.341-2A>G,图3A),使3号内含子的剪切受体由AG变为GG。而先证者无症状的妹妹(Ⅲ-6)以及100名健康对照者均没有发现该突变。
2.3 反转录分析结果 反转录PCR研究表明,c.341-2A>G突变使剪切位点发生改变,其中一条cDNA第4号外显子的前21个碱基(c.341-361)被作为内含子剪切下来(图3B),致使4号外显子mRNA起始的7个氨基酸(p.114-120,即甘氨酸、脯氨酸、天冬氨酸、半胱氨酸、丝氨酸、亮氨酸及脯氨酸)被跨越、未转录。
3 讨论
该家系连续3代均出现发病者,男女均可受累,符合常染色体显性遗传特点。所有患者均在中年发病,主要表现为反复发作的缺血性卒中及认知功能减退;无高血压、糖尿病及高脂血症等脑血管病常见危险因素;头颅MRI显示多发腔梗灶及广泛的脑白质变性;皮肤超微病理检查发现微小动脉平滑肌细胞表面出现GOM沉积,证实CADASIL的诊断。NOTCH3基因突变检测证实存在c.341-2A>G剪切突变,从基因水平进一步确诊了该病。
先证者头MRI改变特点为多发腔梗灶及弥漫性的白质疏松,结合其双侧颞极白质出现长T2信号这一特征性表现,高度提示CADASIL的诊断。研究发现,CADASIL患者的头MRI表现具有一定的演变规律[1],其脑白质病变常早于腔梗灶出现,最早在侧脑室周围及半卵圆中心区域观察到点状或结节状的长T2异常信号,随后病灶增多、融合,形成弥漫性的病灶。该文先证者的白质病变随着病程的延长而逐渐加重,符合这一特点。CADASIL患者的腔隙性梗死灶主要集中在基底节、丘脑及外囊等大脑深部结构,也见于脑室旁白质、皮层下白质、胼胝体及脑干等部位,而本例患者的腔梗灶以侧脑室旁白质及胼胝体为主,基底节病灶相对不明显,提示该病的影像学表现也存在一定的异质性。虽然胼胝体病变罕见于脑小血管病,但在CADASIL患者中并不少见,因此有助于鉴别CADASIL与其他类型的脑小血管病[14]。
NOTCH3基因含有33个外显子,编码含有2321个氨基酸的跨膜蛋白,其胞外段含有34个表皮生长因子重复序列(EGFR),每个EGFR又包括6个半胱氨酸残基[1]。除个别位点外,目前所报道的绝大多数突变均与EGFR中的半胱氨酸残基有关,即突变导致了半胱氨酸残基的生成或丢失,而奇数的半胱氨酸残基打破了原有正常的二硫键的形成,改变了编码蛋白质的构象[13]。本研究中,c.341-2A>G突变位于内含子3的剪切受位,导致NOTCH3基因的mRNA剪切异常,产生了丢失7个氨基酸的截短蛋白,其中包括构成第2个EGFR的第6位半胱氨酸残基(p.117)。该氨基酸丢失后,无法与第108位的半胱氨酸残基配对形成二硫键,进而影响了该EGFR氨基端的双层β折叠的稳定性,使局部结构发生错误折叠而致病[15]。同时该突变具有以下特点:(1)在本家系中与临床表型共分离;(2)国外CADASIL家系中也发现该突变位点[5];(3)NOTCH3基因 mRNA 全长测序没有发现其他致病性突变;(4)100名健康对照者没有发现相同突变。因此综合以上特点支持该突变为致病性突变。
检索文献发现,本家系为目前已知第2个由c.341-2A>G剪切突变所导致的CADASIL家系。第一个家系位于法国东部,其7例受累成员主要表现为各种类型的偏头痛,包括先兆偏头痛、偏瘫型偏头痛、基底动脉型偏头痛以及无先兆偏头痛,仅1例存在认知功能减退[16],随访5年后发现有1例患者出现了缺血性卒中[5],偏头痛常见于高加索CADASIL患者中[10]。而本家系成员则主要表现为脑梗死和痴呆,无明确偏头痛及精神异常病史,与国内CADASIL患者的临床特点一致[10]。由此可见,虽然存在同一个突变位点,但不同种族患者的临床表现可以存在很大差异。目前除个别报道外[17],绝大多数研究均认为CADASIL患者的基因型和临床表型之间并无直接的对应关系[7],而种族及地域因素的差异似乎对CADASIL患者临床表型的影响更大[10,18]。
尽管目前报道的240余种NOTCH3基因突变中仅有3例是剪切突变(除c.341-2A>G外,另2例分别为位于3号内含子深部的c.341-26_24delAAC以及位于15号内含子剪切受位的c.2412-1G>T突变)[5-7]。但研究发现,在人类所有致病性突变中,剪切突变是一种常见的突变类型,其中至少10%的突变为发生在剪切供位或受位的点突变[19],甚至在某些疾病中高达50%的突变是通过影响剪接过程而致病[20]。由于病理证实的CADASIL患者中尚有5%~10%未发现致病性突变[21],因此应该对这些患者进行深入研究,以探讨是否存在NOTCH3基因内含子突变,特别是深在型内含子突变。
[1]Chabriat H,Joutel A,Dichgans M,et al.Cadasil[J].Lancet Neurol,2009,8:643-653.
[2]Tournier-Lasserve E,Joutel A,Melki J,et al.Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12[J].Nat Genet,1993,3:256-259.
[3]O’Sullivan M1,Jarosz JM,Martin RJ,et al.MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL[J].Neurology,2001,56:628-634.
[4]Joutel A,Corpechot C,Ducros A,et al.Notch3mutations in CADASIL,a hereditary adult-onset condition causing stroke and dementia[J].Nature,1996,383:707-710.
[5]Joutel A,Chabriat H,Vahedi K,et al.Splice site mutation causing a seven amino acid Notch3in-frame deletion in CADASIL[J].Neurology,2000,54:1874-1875.
[6]Saiki S,Sakai K,Saiki M,et al.Varicose veins associated with CADASIL result from a novel mutation in the Notch3gene[J].Neurology,2006,67:337-339.
[7]Bianchi S,Dotti MT,Gallus GN,et al.First deep intronic mutation in the NOTCH3gene in a family with late-onset CADASIL[J].Neurobiol Aging,2013,34:2234;e9-12.
[8]Weiming F,Yuliang W,Youjie L,et al.A novel Notch3deletion mutation in a Chinese patient with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy(CADASIL)[J].J Clin Neurosci,2013,20:322-323.
[9]Tan ZX,Li FF,Qu YY,et al.Identification of a known mutation in Notch 3in familiar CADASIL in China[J].PLoS One,2012,7:e36590.
[10]Wang Z,Yuan Y,Zhang W,et al.NOTCH3mutations and clinical features in 33mainland Chinese families with CADASIL[J].J Neurol Neurosurg Psychiatry,2011,82:534-539.
[11]Yin XZ,Ding MP,Zhang BR,et al.Report of two Chinese families and a review of mainland Chinese CADASIL patients[J].J Neurol Sci,2009,279:88-92.
[12]Au KM,Li HL,Sheng B,et al.A novel mutation(C271F)in the Notch3gene in a Chinese man with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy[J].Clin Chim Acta,2007,376:229-232.
[13]王朝霞,吕鹤,张英,等.伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病四个家系的NOTCH3基因突变研究[J].中华神经科杂志,2004,84:1175-1180.
[14]Singhal S,Rich P,Markus HS.The spatial distribution of MR imaging abnormalities in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and their relationship to age and clinical features[J].AJNR Am J Neuroradiol,2005,26:2481-2487.
[15]Dichgans M, Ludwig H, Müller-Höcker J,et al. Small in-frame deletions and missense mutations in CADASIL:3D models predict misfolding of Notch3EGF-like repeat domains[J].Eur J Hum Genet,2000,8:280-285.
[16]Chabriat H,Tournier-Lasserve E,Vahedi K,et al.Autosomal dominant migraine with MRI white-matter abnormalities mapping to the CADASIL locus[J].Neurology,1995,45:1086-1091.
[17]Bianchi S,Rufa A,Ragno M,et al.High frequency of exon 10 mutations in the NOTCH3gene in Italian CADASIL families:phenotypic peculiarities[J].J Neurol,2010,257:1039-1042.
[18]王韵,洪道俊,赵丹华,等.中国26个常染色体显性遗传性脑动脉病伴皮质下梗死和白质脑病家系的临床和影像学特点及与其他国家患者的对比[J].中华神经科杂志,2010,43:697-701.
[19]Krawczak M,Thomas NS,Hundrieser B,et al.Single basepair substitutions in exon-intron junctions of human genes:nature,distribution,and consequences for mRNA splicing[J].Hum Mutat,2007,28:150-158.
[20]López-Bigas N,Audit B,Ouzounis C,et al.Are splicing mutations the most frequent cause of hereditary disease?[J].FEBS Lett,2005,579:1900-1903.
[21]Peters N,Opherk C,Bergmann T,et al.Spectrum of mutations in biopsy-proven CADASIL:implications for diagnostic strategies[J].Arch Neurol,2005,62:1091-1094.
