超高效液相色谱法同时测定减肥类保健食品中非法添加的25种药物
2014-10-22王静文黄湘鹭王钢力张庆生丁丽霞
王静文,黄湘鹭,曹 进*,王钢力*,张庆生,丁丽霞
(1.中国食品药品检定研究院,北京 100050;2.中国药学会,北京 100022)
近年来,随着生活质量的提高,人们对减肥类保健品的需求也越来越大。然而,由于我国保健食品产业发展起步较晚,企业素质参差不齐,市场监督管理体系尚待完善,保健食品因非法添加化学药物而遭到查处的情况屡有报道。作为食品,在保健食品中添加药物的行为不仅违反了《中华人民共和国食品安全法》,而且对消费者的身体健康带来了安全隐患。当前,保健食品中呈现出的非法化学药物添加有如下一些状态:(1)添加物的来源不明,可能添加化学药物包括处方药、现有药物的结构类似物、已下市药物、尚未获得批准的新型药物或先导化合物等[1-4];(2)非法添加药物的剂量随意[1];(3)多种药物添加和多“剂量”水平的添加,由于有部分药物添加处于较低浓度水平,因此,即使检测到,也可能被认为是污染,降低了应受处罚的程度,存在一定的隐蔽性[3];(4)非法添加药物的兼容性不明确,保健食品中非法添加的药物与保健食品配方之间均可能存在相互作用,同时添加的药物也可能与消费者正在服用的药物之间存在相互作用[3]。这些都对消费者的健康存在威胁。
目前用于减肥类保健食品非法添加的筛查方法主 要 有 HPLC 法[4,5]、HPLC-MS 法[6-10]、GC-MS法[11]、薄层色谱法[12]等,但大部分方法都只针对少数几种化合物进行检测,同时检测多种非法添加药物的方法报道较少。本研究依据全国保健食品风险评估报告及相关文献[3-8],选择了25种有减肥作用的化学药物作为研究对象(这些化学药物属于:a.食欲抑制剂(安非他酮、苯佐卡因);b.能量消耗剂(茶碱、咖啡因);c.抗抑郁剂(氟西汀、氢溴酸西酞普兰);d.利尿剂(呋塞米、氢氯噻嗪、螺内酯、布美他尼等);e.胃肠道脂肪抑制剂(瑞莫那班);f.泻药(酚酞、大黄素);g.降糖类(苯乙双胍)),建立了超高效液相色谱(UPLC)分析方法,并进行了方法学验证。通过对17种减肥类保健食品(包括片剂、胶囊、袋泡茶、减肥咖啡等不同制剂的保健食品)的检测,证明本研究建立的方法可有效地应用于减肥类保健品中非法添加化学成分的快速筛查。
1 实验部分
1.1 仪器与材料
ACQUITY超高效液相色谱仪,配有二元溶剂管理器、样品管理器、柱温箱、光电二极管阵列检测器、Empower数据处理系统(美国Waters公司);Milli-Q超纯水器(美国Millipore公司);CF16RXⅡ离心机(日本Hitachi公司);SAS漩涡混合器(英国Stuart公司);KQ3200DE型超声波清洗器(昆山市超声仪器有限公司)。
茶碱、咖啡因、氢氯噻嗪、螺内酯、吲达帕胺、布美他尼、呋塞米、盐酸苯乙双胍、酚酞、大黄素、苄氟噻嗪、盐酸安非他酮、苯丙酸诺龙、苯佐卡因、托拉塞米、氢溴酸西酞普兰、克伦特罗、氯噻酮(纯度均≥98.5%,中国食品药品检定研究院);乙酰唑胺、依尼他酸、三氨蝶啶、氢氟噻嗪、美托拉宗(纯度均≥99.0%,USP);瑞莫那班、唑尼沙胺(纯度均≥99.0%,日本Toronto Research Chemicals公司)。甲醇和乙腈(HPLC级,美国Fisher公司);其他试剂均为分析纯(国药集团化学试剂有限公司)。实验用水为经Milli-Q净化系统过滤的去离子水。
17种减肥类保健食品:包括减肥胶囊、减肥咖啡、减肥茶以及减肥片,来源于市场抽检样品。
1.2 色谱条件
色谱柱:Waters HSS T3柱(100 mm×2.1 mm,1.8 μm);流动相:A.乙腈,B.10 mmol/L 乙酸铵水溶液(含0.1%甲酸);梯度洗脱程序:0~1 min,10%A;1~6 min,10%A~18%A;6~9 min,18%A~28%A;9~12 min,28%A;12~14 min,28%A~35%A;14~20 min,35%A~40%A;20~21 min,40%A ~50%A;21~23 min,50%A ~90%A;23~25 min,90%A;25~25.01 min,90%A ~100%A;25.01~26 min,100%A;26~26.01 min,100%A~10%A;在10%A下平衡2 min,等待下次进样。流速:0.3 mL/min;柱温:40℃;样品室温度:20℃;进样量:2 μL;二极管阵列检测器(DAD)检测波长:200~400 nm,各化合物的检测波长见表1。
1.3 标准溶液的制备
准确称取各种标准品10.0 mg,分别置于10 mL容量瓶中,用甲醇溶解并定容(其中三氨蝶啶用80%的甲醇水溶液定容),配制成1.0 g/L的标准储备溶液,于-4℃(瑞莫那班于-18℃)冰箱中保存备用。临用时,用甲醇稀释上述标准储备溶液,配制成不同浓度的标准工作液。各取单标准储备液适量,以初始流动相为稀释液配制所需浓度的混合标准溶液。
1.4 供试样品的制备
称取供试品适量(约相当于1次用量),研细,置于50 mL离心管中,加入甲醇40 mL,混匀,涡旋1 min,于25℃、100 W 条件下超声15 min,于5000 r/min下离心5 min,取上清液过0.22 μm滤膜,滤液供测定。
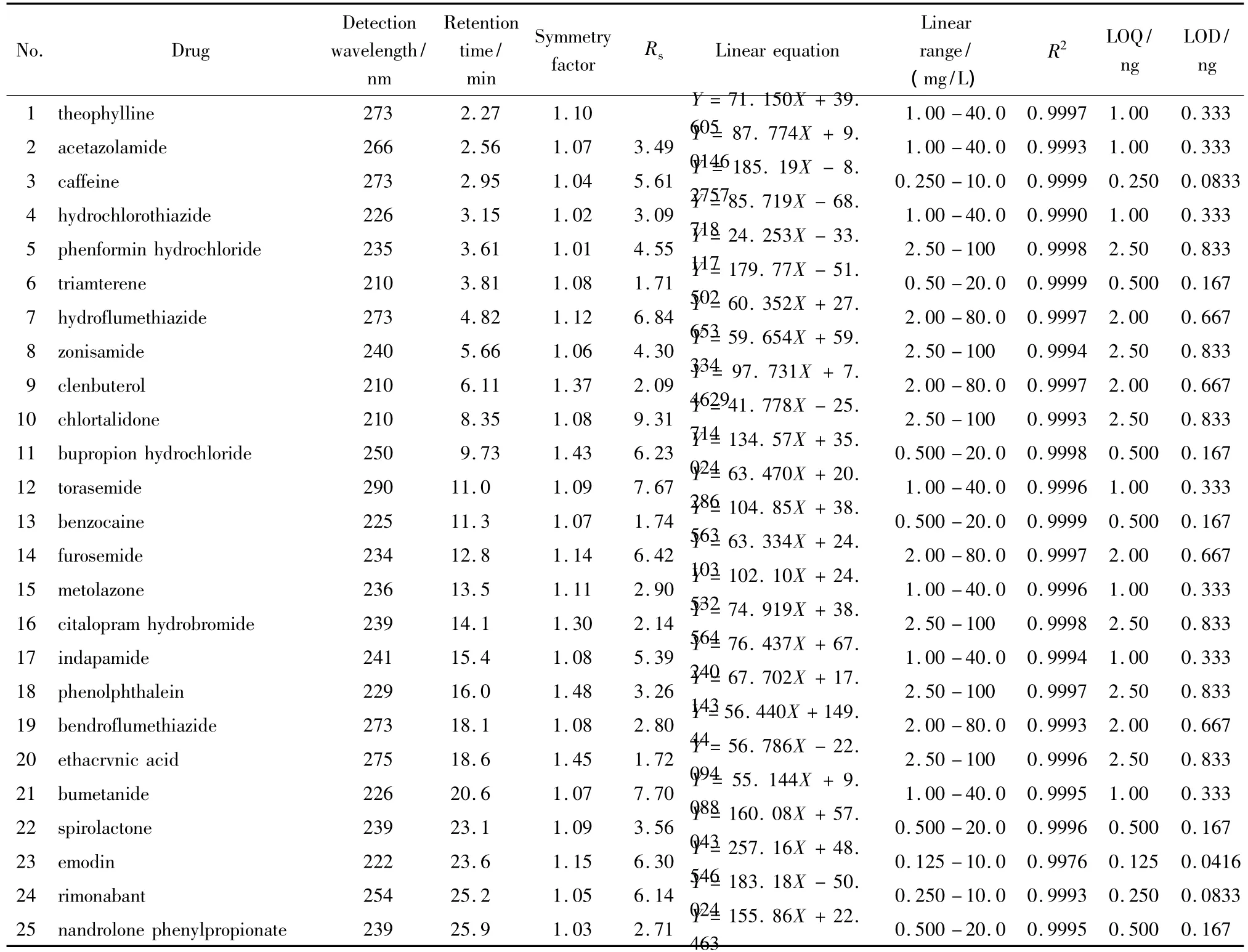
表1 25种化学药物的保留时间、对称因子、分离度、线性方程、线性范围、相关系数、定量限和检出限Table 1 Retention times,symmetry factors,resolutions(Rs),linear equations,linear ranges,correlation coefficients(R2),LOQs and LODs of the 25 drugs
2 结果与讨论
2.1 样品提取方法的优化
为了减少操作步骤及获得尽可能高的提取效率,本文优化了超声提取方式。分别考察了不同提取溶剂(甲醇、乙腈、水)和不同提取时间(10、15、20、25、30 min)等条件下的提取效果。结果表明,以甲醇为提取溶剂、超声提取15 min的提取效率最高,待测定成分提取完全。
2.2 色谱条件的选择
2.2.1 色谱柱的选择
待分离的 25 种化合物中,文献[4-8,13-18]均采用C18反相色谱柱进行分离,所以本实验对UPLC系统中常用的C18色谱柱Waters BEH C18柱(100 mm ×2.1 mm,1.8 μm)和 Waters HSS T3柱(100 mm×2.1 mm,1.8 μm)进行了考察。结果显示两种色谱柱均存在难分离物质对,均需要对液相色谱条件进一步改善,但使用Waters HSS T3柱时化合物的理论塔板数普遍高于使用Waters BEH C18柱时,峰形较尖锐,Waters HSS T3的填料是以高纯硅胶作为载体,机械强度较高,可以耐受高压,适合于UPLC的使用,其碳载量为11%,小于BEH C18的17%,有利于极性较大的化合物分析,故选用Waters HSS T3进行进一步的色谱条件优化。
2.2.2 流动相的选择
由于待分析的化合物种类较多,极性差别较大,梯度洗脱更有利于分离。文献[4-8,13-18]中流动相条件差别较大,如以0.1 mol/L磷酸二氢钠-乙腈(pH 3.0±0.1)为流动相测定氢氯噻嗪含量[13],以甲醇-乙腈-水为流动相测定唑尼沙胺含量[14],以己烷磺酸钠柠檬酸溶液-乙腈为流动相测定酚酞含量[15];有的一种化合物有多种流动相条件[13,16],也有些化合物没有可参考的液相色谱条件。鉴于以上情况,本实验首先对乙腈-水二元流动相系统进行考察,发现分离效果较差,美托拉宗/西酞普兰、苄氟噻嗪/依尼他酸等化合物的峰重合,乙酰唑胺、氢氯噻嗪、酚酞等化合物峰形较差,出现双头峰、拖尾峰等现象,调节流动相pH使其偏酸性时峰形得到改善(推测中性条件下氢氯噻嗪、乙酰唑胺等化合物发生电离,同时以分子与离子两种形式存在,出现二次分配,使得色谱峰分叉),这与大多数文献中使用偏酸性流动相条件相符。进一步考察0.15%三氟乙酸(pH 1.8)、10 mmol/L乙酸铵(含 0.1%甲酸,pH 3.3)、10 mmol/L乙酸铵(乙酸调pH 4)、10 mmol/L磷酸二氢钠(磷酸调pH 5)这4种流动相条件对25种化合物的分离情况,结果表明:当pH≥4时,部分化合物仍出现双头峰;当pH=1.8时,大黄素出现双峰,大黄素属于蒽醌类化合物,在酸性条件下易被还原,生成蒽酚及其互变异构体蒽酮;在pH=3.3条件下,25种化合物得到较好的分离,同时乙酸铵为挥发性缓冲盐,便于后续研究中实现液相色谱到液相色谱-质谱方法的转变。
2.2.3 柱温的选择
研究发现,色谱柱的柱温对上述难分离物质对唑尼沙胺/克伦特罗和苄氟噻嗪/依尼他酸这两对色谱峰之间的分离度有显著的影响,本实验对不同柱温条件(30、35、40、45℃)进行了考察。结果表明,在40℃时,两难分离物质对的分离效果最佳,同时其他物质保持良好的分离状态。
2.2.4 色谱峰的定性
本文主要根据对照品的保留时间以及相应峰位的紫外扫描图对25种化学药物色谱峰定性。25种化学药物的保留时间见表1,254 nm波长下的液相色谱图见图1。
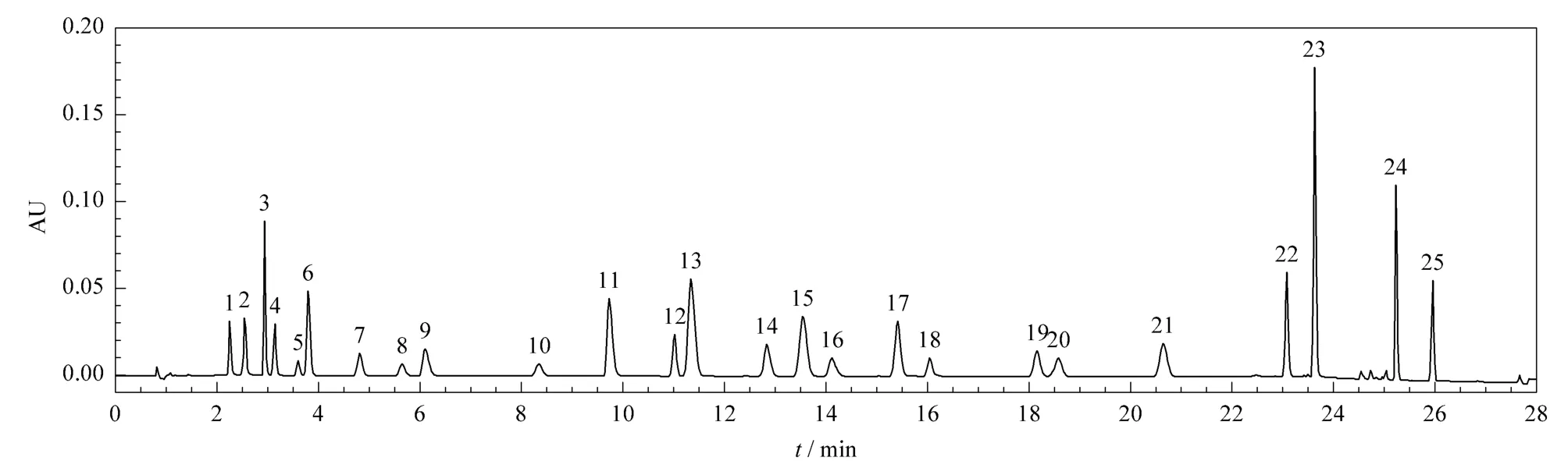
图1 254 nm下25种化学药物的UPLC谱图Fig.1 UPLC chromatogram of the 25 drugs at 254 nm
2.3 线性关系、定量限与检出限
吸取一定量的单标准储备液,用初始流动相稀释,配制成一系列不同浓度的混合标准溶液;按1.2节的条件进样分析,以峰面积Y对对照品的质量浓度X(mg/L)进行回归分析,按照S/N=10计算定量限(LOQ),按照S/N=3计算检出限(LOD),25种化学药物的回归方程、线性范围、相关系数、定量限及检出限见表1。
2.4 方法的回收率和精密度
在经测定不含有25种化学药物的空白样品(苹果醋减肥胶囊)中进行加标回收率和精密度试验。在空白样品中添加低、中、高3个水平的标准溶液,按本方法进行实验,平行测定6次,以评价该方法的准确度。所得到的平均加标回收率范围为70.7%~104%,相对标准偏差(RSD)均不大于5.03%(见表2)。
2.5 实际样品的测定
应用本方法对17种减肥类保健食品进行了分析测定,根据欧盟委员会非强制执行法案2002/657/EC的规定,通过比较样品与标准品保留时间及色谱峰3个峰位点的紫外吸收特征扫描光谱图定性,结果如下:3种样品中检测出酚酞,含量分别为8.54、0.509、3.87 mg/g;1种样品中检测出大黄素,含量为0.0648 mg/L(该样品标签中不含有大黄素类组分),未超出中华人民共和国药典(一部)中规定的用量[18]。
酚酞属于联苯甲烷衍生物类泻药,根据国家食品药品监督管理局药品检验补充检验方法和检验项目批准件2006004和食药监办许[2010]114号文件,酚酞为减肥类保健食品中的违禁添加物;大黄素属于蒽醌类衍生物类泻药,尽管蒽醌类衍生物可用于保健食品中,但使用量应有所控制。长期服用这两种药物会带来严重的不良反应,服用酚酞会诱发顽固性便秘、肠炎皮疹、大出血等;服用蒽醌类泻药会导致大肠黑便病,增加肝毒性,甚至有致畸和致癌等作用[19]。
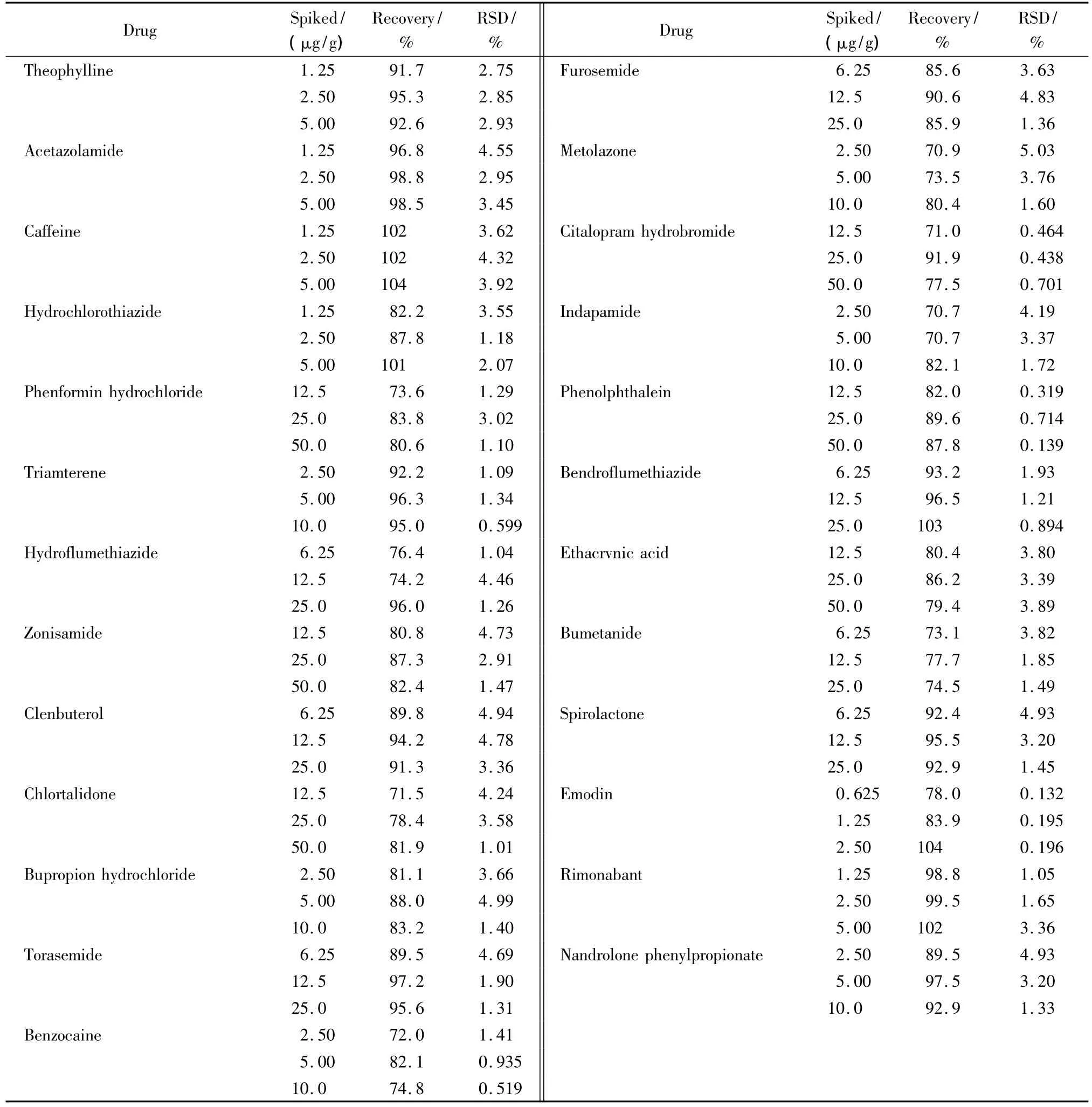
表2 空白样品中25种化学药物的添加回收率及精密度(n=6)Table 2 Recoveries of the 25 drugs spiked in a blank matrix(n=6)
3 结论
本文建立了使用同一种色谱条件同时测定减肥类保健食品中可能添加的25种减肥化学药物含量的UPLC方法,在26 min内即完成了对各化合物的分析测定。与传统液相色谱方法相比,在保证分离度的情况下,实现了快速有效的高通量分析,检测的化合物种类较为全面,可为监测保健食品非法添加药物提供强有力的技术支持。所采用的液相色谱条件相对简单,便于实现液相色谱法到液相色谱-质谱法的转换,可以进行不同方法间的相互验证。同时,完成一次样品分析流动相的消耗量仅为7.8 mL,既降低了成本,又减少了废液排放,具有重要的环保意义。
[1]Lee H M,Kim C S,Jang Y M,et al.J Pharm Biomed Anal,2011,54:491
[2]Lee E S,Kim J W,Lee J H,et al.Food Addit Contam:Part A,2013,30(4):621
[3]Tang M H Y,Chen S P L,Ng S W,et al.Br J Clin Pharmacol,2010,71(2):250
[4]Rebiere H,Guinot P,Civade C,et al.Food Addit Contam:Part A,2012,29(2):161
[5]Deconinck E,Verlinde K,Courselle P,et al.J Pharm Biomed A-nal,2012,59:38
[6]Li Y,Hu J T,Shi Y,et al.Food Anal Method,2011,4:505
[7]Ma W,Peng T,Zhu M D,et al.Chinese Journal of Analytical Chemistry(马微,彭涛,朱明达,等.分析化学),2009,37(11):1583
[8]Ma W,Ma Q,Fu L,et al.Chinese Journal of Chromatography(马微,马强,付丽,等.色谱),2010,28(1):43
[9]Zhu L,Ruan L P,Liu H L,et al.Chinese Journal of Chromatography(朱琳,阮丽萍,刘华良,等.色谱),2013,31(7):709
[10]Wang M L,Yan H F,Fu S L,et al.Chinese Journal of Chromatography(王美玲,颜鸿飞,傅善良,等.色谱),2012,30(10):980
[11]Li Y,Zhang H,Hu J T,et al.J Chromatogr Sci,2012,50(10):928
[12]Shi Y Q,Yao J,Zhang Q M,et al.Chinese Journal of Pharmaceutical Analysis(施亚琴,姚静,张启明,等.药物分析杂志),2007,27(1):36
[13]Pharmacopoeia Commission of People’s Republic of China.Pharmacopoeia of People’s Republic of China,Part 2.Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典,二部.北京:中国医药科技出版社),2010:513,555
[14]Bao C G,Yang Y L,Yu Y B,et al.Pharmaceutical Journal of Chinese People’s Liberation Army(包存刚,杨永林,余以兵,等.解放军药学学报),2010,26(1):70
[15]Ge B K,Zhang Q Q,Zhao K X.Chinese Journal of Pharmaceutical Analysis(葛宝坤,张群谦,赵孔祥.药物分析杂志),2012,32(11):2034
[16]Sun Y Y,Wei Y,Wang K S,et al.Chinese Journal of Pharmaceutical Analysis(孙莹莹,韦阳,王恪申,等.药物分析杂志),2010,30(3):396
[17]Pharmacopoeia Commission of People’s Republic of China.Pharmacopoeia of People’s Republic of China,Part 1.Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典,一部.北京:中国医药科技出版社),2010:22
[18]Ma W,Wang H B,Chen D D,et al.Food Science and Technology(马微,王海波,陈冬东,等.食品科技),2010,35(8):339
