烟嘧磺隆分子印迹微球的制备及其吸附性能的考察
2014-10-22赵尔成贾春虹朱晓丹
夏 英,张 澜,*,赵尔成,贾春虹,朱晓丹
(1.大连工业大学纺织与材料学院,辽宁 大连 116034;2.北京市农林科学院植物保护环境保护研究所,北京 100097)
磺酰脲类除草剂是20世纪70年代发展起来的一类高效、低毒的新型除草剂。其因生物活性高、用量少、在环境中容易降解而成为除草剂市场的热点。烟嘧磺隆(NS)是磺酰脲类除草剂中的一种,也得到了广泛的使用。但随着该类除草剂使用量及使用范围的不断加大,其在农作物中的残留、对环境的污染及对人体健康造成的危害日益引起人们的重视[1]。当前,用来分析磺酰脲类除草剂残留的方法主要有高效液相色谱(HPLC)[2,3]、毛细管电泳(CE)[4]和高效液相色谱-质谱(LC-MS)[5,6]方法。但是该类除草剂的性质不稳定且残留量低,因此在检测前需要进行提取和富集。而传统的前处理技术通常是使用乙腈或磷酸盐缓冲液进行提取,再经过C18小柱或弗罗里硅土小柱进行净化,由于这些小柱并没有特异性的吸附能力,因此其净化、富集效果并不理想。
由于分子印迹聚合物含有一个与模板分子(目标化合物)在空间结构上完全匹配,并能与模板分子专一结合的功能基的三维空穴,因此分子印迹聚合物对目标化合物具有专一性的识别作用[7-9]。李兆敏等[10]以烟嘧磺隆为模板分子,采用本体聚合法成功地制备了烟嘧磺隆分子印迹聚合物,然而本体聚合法制备的分子印迹聚合物往往成棒状或块状,需要对其进行研磨筛分,不仅后处理过程繁琐,且极易破坏聚合物的分子结构。本文选择以氯仿为溶剂、甲基丙烯酸(MAA)为功能单体,采用沉淀聚合法制备烟嘧磺隆分子印迹微球,对其制备工艺进行了优化,考察了NS和MAA之间的作用机理,并采用高效液相色谱及固相萃取(SPE)等手段对微球的吸附性能进行了研究。
1 实验部分
1.1 试剂与仪器
烟嘧磺隆(中国农业大学农药分析实验室);吡嘧磺隆(PS)、氯嘧磺隆(CMS)、甲磺隆(MSM)标准储备液及MAA、丙烯酰胺(AM)、三甲氧基丙烷三甲基丙烯酸酯(TRIM)(J&K公司);偶氮二异丁腈(AIBN)(上海国药集团);乙腈、甲醇、醋酸(色谱纯,迪马公司),氯仿、二氯甲烷(分析纯,北京化工厂)。去离子水(Milli-Q纯水机)。
日本岛津公司SCL-10Avp型高效液相色谱、SIL-10AP自动进样器、SPD-M10Avp二极管阵列检测器;DU800型紫外分光光度计(美国BECKMAN公司);Sartorius电子天平(1/1000,北京赛多利斯天平有限公司);ZHQ型磁力平底电热套(北京瑞成伟业仪器设备有限公司);DHG-9075A型电热恒温鼓风干燥箱(上海一恒科技有限公司)。
1.2 HPLC 条件
色谱柱:Agilent ZORBAX ODS柱(250 mm×4.6 mm,5 μm);流动相:乙腈/水/醋酸(40∶60∶0.2,v/v/v);流速:1.0 mL/min;检测波长:254 nm;柱温:30℃;进样量:10 μL。
1.3 烟嘧磺隆分子印迹聚合物的制备
以NS为模板分子、MAA为功能单体、TRIM为交联剂、AIBN为引发剂、氯仿为致孔剂,通过沉淀聚合法合成了烟嘧磺隆分子印迹聚合物微球(MIPMs)。
称取0.5 mmol的NS溶于90 mL的氯仿中,加入2 mmol的MAA,在转速为150 r/min的恒温振荡器下振荡4 h后,放入4℃冰箱中过夜形成预聚合物。向其中加入2 mmol的 TRIM和0.5 mmol的AIBN,超声波脱气10 min后,充氮气10 min。密封后,在60℃热引发下聚合24 h,再将混合液冷却、离心,得到制备的聚合物。聚合物经含20%(v/v)乙酸的甲醇溶液洗涤至无模板分子为止,再用甲醇洗掉残留的乙酸,干燥,即得NS-MIPMs。
空白印迹聚合物微球(NIPMs)的制备除不加模板分子外,其他步骤均与印迹聚合物的制备相同。
1.4 紫外光谱分析
固定MAA的浓度,改变NS的浓度:分别向0.1 mmol/L的 MAA 中 加 入 0.018、0.038、0.075 mmol/L的NS,配制一系列的混合溶液,在振荡器上振荡4 h后,放在冰箱中过夜,再以含有对应浓度NS的乙腈溶液为参比扫描吸收光谱,在适当波长下测得吸光度。
固定NS的浓度为0.01 mmol/L,渐增MAA的浓度(0.01~0.08 mmol/L),配制一系列的混合溶液,并在振荡器上振荡4 h后,放在冰箱中过夜,再以乙腈溶液为参比测定吸光度;同时配制一系列相同的混合溶液,不经过振荡直接使用紫外分光光度计测定吸光度,考察模板分子和功能单体在溶剂中反应前后的吸光度差。
1.5 聚合物微球的静态吸附实验
取一组等量的NS-MIPMs和NIPMs各10 mg于2 mL的离心管中,再分别加入1.5 mL不同质量浓度(3~125 mg/L)NS的二氯甲烷溶液。将混合液放到25℃恒温振荡器中振荡24 h后,将吸附液离心5 min并过膜,取一定量的吸附液用氮气吹干后,用乙腈溶解并定容,供HPLC分析。
1.6 聚合物微球的吸附动力学研究
固定NS的质量浓度为100 mg/L,分别向其加入等量的MIPMs和NIPMs各10 mg,放到25℃恒温振荡器振荡不同时间(5~120 min)后,将吸附液离心5 min并过膜,取一定量的吸附液用氮气吹干后,用乙腈溶解并定容,供HPLC分析。
1.7 聚合物微球的选择性吸附研究
配制 50 mg/L 的 NS、CMS、MSM、PS的混合标准溶液待用;取一组等量的NS-MIPMs和NIPMs各10 mg于2 mL离心管中,再分别加入1.5 mL上述混合标准溶液。将混合标准溶液放到25℃的恒温振荡器中振荡24 h后,将吸附液离心5 min并过膜;取一定量的吸附液用氮气吹干后,用乙腈溶解并定容,供HPLC分析。
1.8 NS-MIPMs固相萃取小柱的制备
准确称取100 mg制备的NS-MIPMs和NIPMs,分别填充于聚丙烯固相萃取小柱空管(60 mm×10 mm)中,预先在底端放入一层聚乙烯筛板,压紧聚合物,使其表面平齐,上面再加入一层筛板,用玻璃棒推挤筛板压实聚合物,继续轻敲小柱,直至筛板与聚合物微球间紧密结合。
1.9 MISPE-HPLC检测土壤中烟嘧磺隆的前处理方法
准确称取土壤样品10.00 g(精确至0.01 g)置于50 mL聚四氟乙烯离心管中,加入5 mL磷酸盐缓冲液(pH 7.8),涡旋1 min后,加入10 mL含2%(v/v)甲酸的乙腈提取液,涡旋提取5 min,加入2 g NaCl和4 g无水 MgSO4,立即涡旋1 min,于4000 r/min下离心5 min,取2 mL上层清液待MISPE净化。
取制备好的NS-MIPMs固相萃取小柱,先用5 mL二氯甲烷活化小柱,接着加入2 mL上述提取液到小柱中,再用4 mL二氯甲烷对小柱进行淋洗,最后用1 mL二氯甲烷/乙腈(2∶1,v/v)混合溶液洗脱3次,收集洗脱液并蒸发至干,用流动相溶解并定容至1 mL,供HPLC分析。
2 结果与讨论
2.1 分子印迹聚合物的制备
在制备分子印迹聚合物微球的过程中,不同的制备工艺会使分子印迹微球的吸附能力有所不同,因此为了使聚合物具有最佳的吸附性能,本文对烟嘧磺隆分子印迹聚合物的制备工艺进行了优化,考察了功能单体种类、致孔剂种类、致孔剂体积、模板分子与功能单体的物质的量比以及模板分子与交联剂的物质的量比等因素对聚合物吸附性能的影响(见表1)。
根据M1~M3聚合物吸附能力的比较,我们选择使用MAA作为功能单体、氯仿作为反应溶剂来制备烟嘧磺隆分子印迹聚合物。而根据M1和M4~M8的比较发现,增加致孔剂的体积有助于提高MIPMs的吸附能力,同时用扫描电镜对其进行形貌考察(见图1),当致孔剂体积为50、60 mL时形成的聚合物并不呈球形而是相互团聚成块状;当致孔剂体积为70 mL时,有部分聚合物以微球形式析出,但仍有团聚现象;而当致孔剂体积增加到80、90 mL时,形成的聚合物全部以微球形式析出,且90 mL时析出的微球体积最小。这可能是由于当致孔剂的体积变大时,在聚合反应中聚合物从溶液中析出后彼此之间的距离增大而不易发生团聚,且悬挂在聚合物表面上的交联剂的残余双键也较难再次捕捉单体,因此使得形成的聚合物微球体积小、比表面积大,具有较好的吸附能力[11,12]。但当溶剂量达到100 mL时,吸附量增加并不明显,且产量极低,这可能是因为溶剂量过多会导致交联剂与功能单体之间距离过大而无法聚合生成聚合物,因此本文选择使用的致孔剂体积为90 mL。对M1和M9~M13进行比较,考察功能单体和交联剂的用量对聚合物吸附性能的影响。通常当功能单体用量过少时,会形成较少的识别位点;而用量过多时,则功能单体会趋向于自聚合形成较多的非特异性分子印迹聚合物而影响吸附能力。对于交联剂,同样需要合适的用量,交联剂使用量过多会导致聚合物过于坚硬而不易洗掉模板分子;而用量过少则聚合物的耐溶剂性差,在反应中识别位点易被破坏,导致吸附能力变差[13]。从表1可以看到,当模板分子/功能单体/交联剂/引发剂的物质的量比为1∶4∶4∶1时,制备的 NS-MIPMs的吸附能力最好,因此最终选择制备烟嘧磺隆分子印迹微球的工艺为NS/MAA/TRIM/AIBN的物质的量比为 1∶4∶4∶1,致孔剂为90 mL 的氯仿。
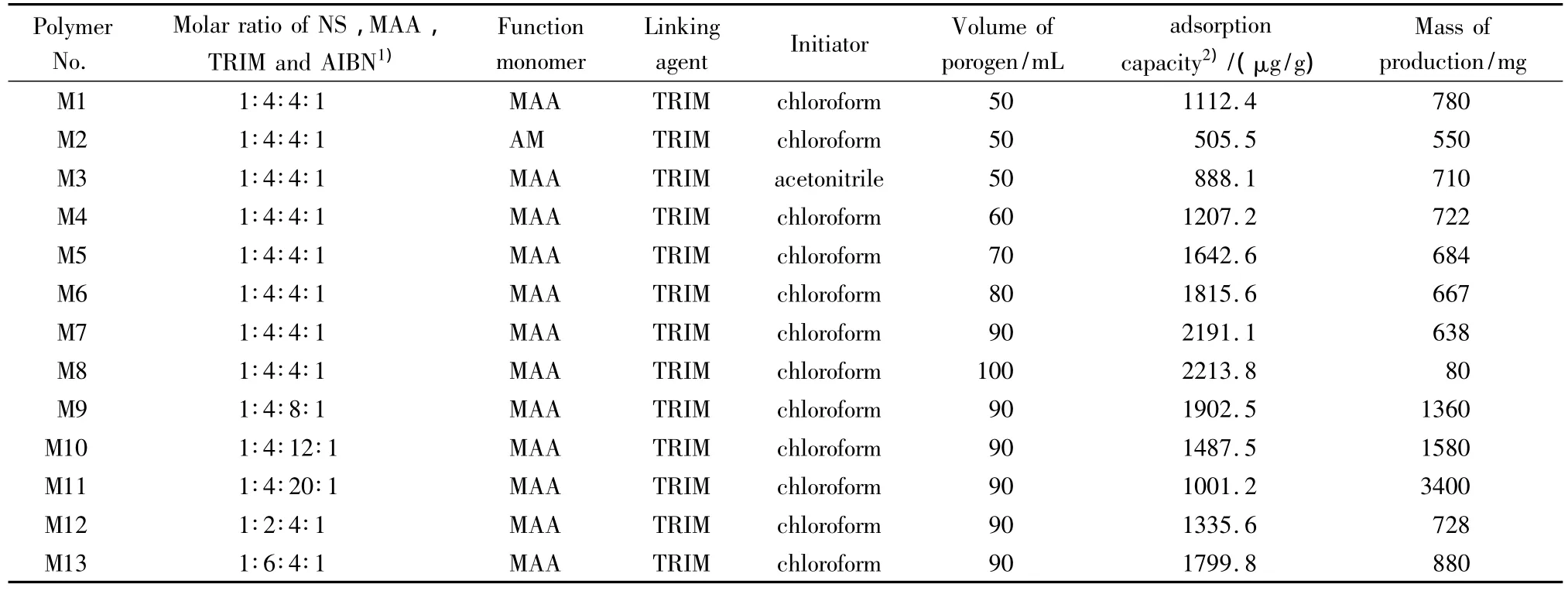
表1 预聚合物的组成及对应MIPMs的吸附量Table 1 Composition of prepolymerization mixtures and adsorption efficiency for MIPMs

图1 不同溶剂量下制备的MIPMs的形貌图Fig.1 Scanning electron micrographs of NS-MIPMs
2.2 NS和MAA的作用机理研究
在非共价键法制备分子印迹聚合物中,模板分子和功能单体之间能否通过某种非共价键力结合在一起是决定分子印迹聚合物亲和性能的关键因素。通常,当模板分子和功能单体之间存在相互作用时,会由于质子供体的贡献导致X-H的键略有伸长或收缩效应,因此在紫外分光光度检测时,混合溶液的紫外吸收会发生红移或蓝移现象,且吸光度也会发生改变[14]。本文固定MAA浓度为0.1 mmol/L,逐渐增加NS的浓度(从0.018~0.075 mmol/L),采用紫外分光光度计测得混合溶液的吸收曲线(见图2a)。从图2a中可以看到,随着NS浓度的增加,混合溶液的最大吸收波长发生蓝移,吸光度也随之增大;而NS和MAA的吸光度都相对较低,说明混合溶液吸光度的变化并不是由于NS的背景效应所引起的,因此证明NS和MAA之间的确存在某种非共价键力使两者连接在一起[15]。
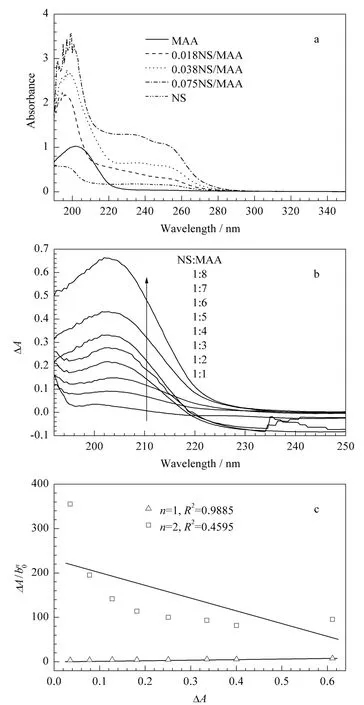
图2 (a)MAA、NS及MAA和NS混合溶液的紫外吸收光谱、(b)NS和MAA混合溶液的紫外吸光度差光谱(混合溶液中 NS 与MAA 的物质的量比分别为1∶1,1∶2,1∶3,1∶4,1∶5,1∶6,1∶7,1∶8)、(c)在202 nm 下ΔA/bn0 对ΔA作图Fig.2 (a)absorption spectra of MAA,NS and MAA in the presence of various amounts of NS,(b)absorption spectra of the differential absorbance of NS in the presence of MAA(the molar ratio of NS to MAA was 1∶1,1∶2,1∶3,1∶4,1∶5,1∶6,1∶7,1∶8,respectively)and(c)plot of ΔA/bn0versus ΔA at 202 nm
固定NS浓度为0.01 mmol/L,逐渐增加MAA的浓度(从0.01~0.08 mmol/L),在紫外分光光度计上测得两者在溶剂中作用前后的吸收曲线,从而绘制出紫外吸光度差光谱(见图2b)。
在理论[14]上,模板分子与功能单体形成复合物,可以用式(1)来表示。
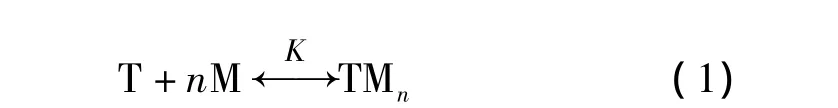
式中K是结合常数;n=1,2,3,…;T代表模板分子;M代表功能单体。以a0表示模板分子的浓度,b0表示功能单体的浓度,因此复合物的浓度c可推导为式(2)。
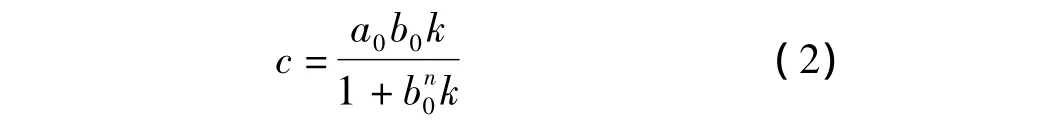
由于功能单体与模板分子以及其主客体复合物最大吸收波长在202 nm处,所以功能单体与模板分子在乙腈溶液中作用前后的吸光度差ΔA可表示为式(3)。
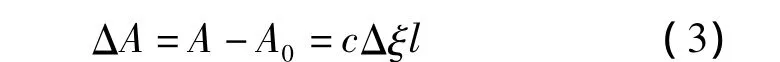
式中,l为光在样品中经过的距离,ξ为吸收系数,Δξ=ξC- ξA- ξB(其中 ξC、ξA、ξB分别代表复合物、模板分子与功能单体的吸收系数)。由此可得式(4)。
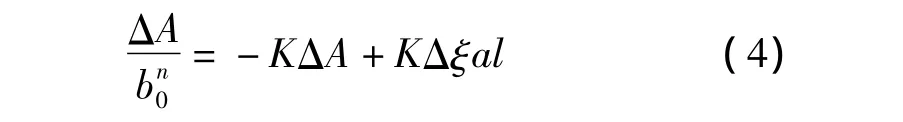
分别将n=1和n=2带入式(4),得图2c,从图2c中可以看到当n=1时,以对ΔA作图是一条直线(R2=0.9885);而当n=2时,所得到的数值并不呈线性。这说明在考察的浓度范围内,模板分子与功能单体的相互作用形式主要为一个NS与一个MAA发生作用[16]。推测为 NS中的-NH和 MAA中的-COOH以氢键相结合,作用机理如图3所示。
2.3 平衡吸附实验及Scatchard分析
通常采用静态平衡吸附法来考察分子印迹聚合物对模板分子的亲和性能,图4a为NS-MIPMs和NIPMs的等温吸附曲线。分子印迹聚合物的吸附量可根据式(5)求得。
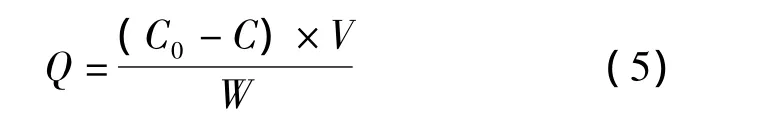
式中:Q为吸附平衡时聚合物的吸附量(μg/g);C0和C分别为吸附前和吸附后溶液中NS的浓度(μg/mL);V为吸附液的体积(mL);W为印迹聚合物的质量(g)。从图4a中可以看到,随着溶液中NS浓度的逐渐增大,NS-MIPMs和NIPMs的吸附量也随之提高,而且 NS-MIPMs的吸附量明显高于NIPMs的吸附量。这是由于在制备NS-MIPMs的过程中,NS和MAA之间通过非共价键力的作用连接在一起,并在交联剂和引发剂的作用下形成聚合物,当除去聚合物中的模板分子时,就形成了能与模板分子形状相匹配且具有特异识别位点的三维空穴,因此NS-MIPMs对模板分子NS具有较高的吸附能力[17]。而NIPMs则是功能单体的随机聚合,对NS只有非特异性吸附,所以吸附能力相对较差。
图3 NS和MAA之间的作用机理Fig.3 Schematic diagram of the interaction between NS and MAA
Scatchard一直是分析分子印迹聚合物吸附性能的经典方法。它是通过将竞争结合曲线转换成直线,从而得到印迹聚合物对模板分子结合位点的个数以及聚合物的最大表观结合量。图4b为根据Scatchard模型对NS-MIPMs的结合特性分析得出的结论,可由式(6)得到。
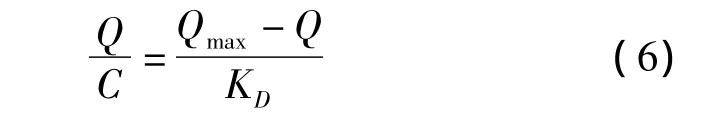
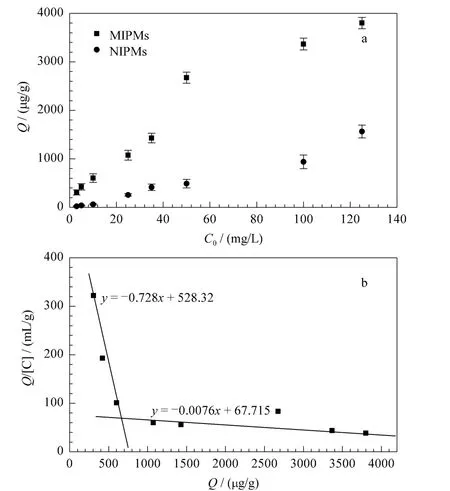
图4 (a)NS-MIPMs和NIPMs对NS的等温吸附曲线和(b)NS-MIPMs的Scatchard分析曲线Fig.4 (a)Binding isotherms of NS on NS-MIPMs and NIPMs and(b)Scatchard plot of NSMIPMs for estimating the binding nature
式中:Q为吸附平衡时NS-MIPMs对NS的结合量(μg/g),C为吸附平衡时溶液中游离的NS的浓度(μg/mL),Qmax为最大表观结合量(μg/g),KD为结合位点的解离常数。从图4b中可以看到,Scatchard分析图有两个明显不同的区域,对其进行线性回归得到回归方程分别为y=-0.728x+528.32和y=-0.0076x+67.715,这说明在预聚合过程中,模板分子和功能单体之间的结合是不均匀的,存在两类结合位点。推测原因可能是由于在聚合中,加热易造成溶液中热力学条件的不均匀,导致模板分子和功能单体之间形成了不同组成的复合物,同时由于自由基聚合本身的不可控性,聚合中形成的空穴大小不同也会使得印迹聚合物对印迹分子的识别具有非均一性[18]。其中,高亲和性结合位点的解离常数KD1和最大结合量Qmax1分别为158.73和11370.5 μg/g;低亲和性结合位点的解离常数KD2和最大结合量 Qmax2分别为1.37 和725.7 μg/g。
2.4 分子印迹聚合物微球的吸附动力学研究
为了进一步研究分子印迹微球对烟嘧磺隆的吸附性能,并考察其对烟嘧磺隆的最佳吸附时间,我们对NS-MIPMs和NIPMs进行了吸附动力学研究。将一定量的NS-MIPMs和NIPMs分别加入到固定浓度的NS溶液中,振荡吸附不同时间后,通过静态平衡吸附法测得不同时间下NS-MIPMs和NIPMs对NS的吸附量,并以吸附量Q对吸附时间做动力学吸附曲线,如图5所示。从图5中可看到,分子印迹微球对模板分子的吸附主要发生在前30 min,随后吸附量增加缓慢,当吸附时间达到1 h时,吸附量几乎达到饱和。由此推测,聚合物中带有识别位点且能与模板分子相互匹配的三维空穴大都存在于聚合物的表面,且形成的空穴体积较大[19],因此可以快速地吸附目标化合物。同时仍可观察到,NS-MIPMs对NS的吸附量要高于NIPMs,再一次证明在制备NSMIPMs过程中,成功地形成了与NS形状相匹配的三维空穴,实现了对NS的特异性吸附。
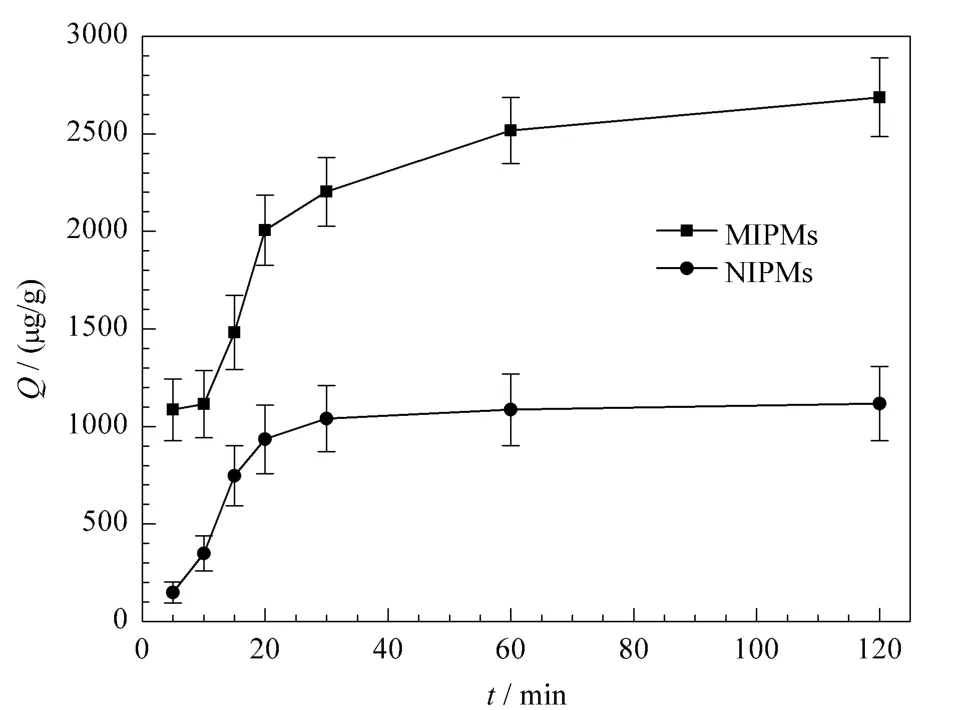
图5 NS-MIPMs和NIPMs的吸附动力学曲线Fig.5 Adsorption dynamic curves of the NS-MIPMs and NIPMs
2.5 分子印迹微球的选择性吸附研究
为考察分子印迹聚合物的特异性吸附能力,就NS-MIPMs和NIPMs对NS及其结构类似物(MSM、CMS、PS)的吸附性能进行了测试。图6为 NSMIPMs及NIPMs在相同时间范围内对4种化合物的吸附量比较,可以看到,NIPMs对NS及其结构类似物的吸附量都小于NS-MIPMs,但无论NS-MIPMs还是 NIPMs均对 NS的吸附量最大,而对 MSM、CMS、PS 3种化合物的吸附较差。这是因为在制备NS-MIPMs时是以NS为模板,当去除模板分子后形成洞穴的空间结构与识别位点都与NS匹配,所以NS-MIPMs可以快速捕捉并吸附目标分子NS。而对于NS的结构类似物,虽然其分子结构上存在能与NS-MIPMs相识别的位点,但是空间结构也是影响聚合物特异性吸附能力的重要因素之一,因此MSM、CMS、PS 3种化合物只能与NS-MIPMs的部分识别位点相互作用,所以吸附量较低,这说明以NS为模板分子制备的分子印迹聚合物对NS具有较好的专一性吸附能力。
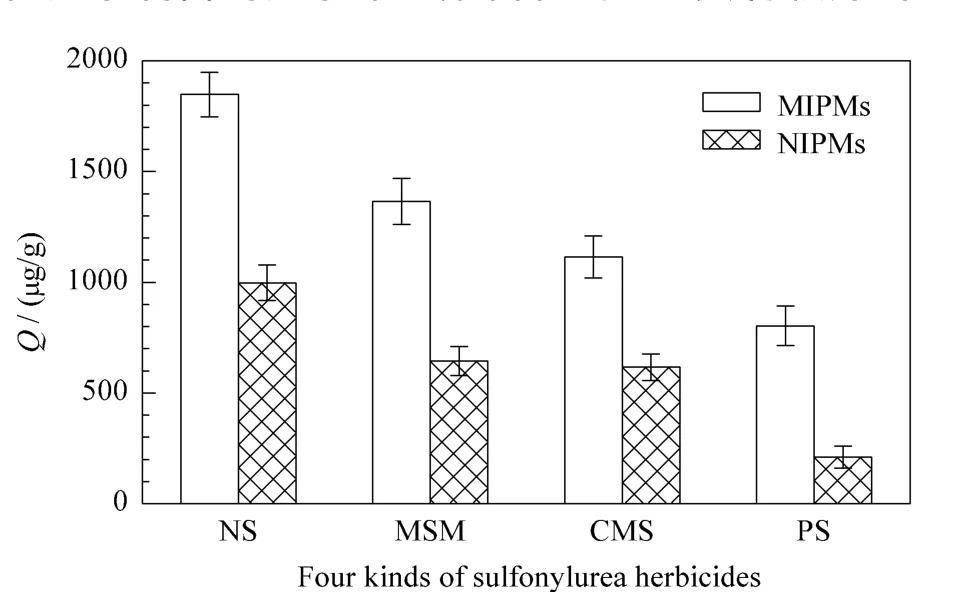
图6 NS-MIPMs及NIPMs对NS及其结构类似物的吸附选择性Fig.6 Adsorption selectivity of NS-MIPMs and NIPMs for NS and its structure analogues
2.6 分子印迹聚合物微球在SPE小柱中的应用
由于分子印迹聚合物微球对模板分子存在特异性吸附,因此常被应用于固相萃取技术中来实现对目标化合物的痕量检测。本文也将合成的NSMIPMs应用于固相萃取技术中,制备了烟嘧磺隆分子印迹固相萃取小柱NS-MISPE,优化了淋洗条件,建立了NS-MISPE-HPLC检测土壤中烟嘧磺隆残留量的分析方法。
2.6.1 NS-MISPE 淋洗条件的优化
对活化溶液和上样溶液进行优化不仅可以保证小柱的保留性能,而且可以增强聚合物内识别位点的活性。通常选择聚合反应时的致孔剂作为活化溶液和上样溶液,这样可以使MISPE对目标化合物具有最佳的吸附性能[20]。但是溶剂的极性同样影响聚合物对模板分子的亲和能力,在分子印迹聚合物吸附模板分子的过程中,不同位置的识别位点与模板分子之间非共价键力的强弱不同,而弱极性溶剂会较少地破坏非共价键力的形成,因此可以保证MISPE对目标化合物的保留性能。常用的上样溶液有乙腈、二氯甲烷、氯仿、丙酮、水等,其极性分别为6.2、3.4、4.4、5.4、10.2。其中以二氯甲烷的极性为最低,而本实验采用的致孔剂为氯仿,因此我们分别采用2 mL二氯甲烷和2 mL氯仿作为上样溶液进行对比试验。结果表明,采用二氯甲烷作为上样溶液可以使目标化合物完全保留在固相萃取小柱内,而采用氯仿为上样溶液则会有一小部分目标化合物随溶液流出柱外。因此本文选择二氯甲烷作为活化溶液和上样溶液,用量分别为5 mL和2 mL。
淋洗溶剂的选择对于保证MISPE的回收率具有至关重要的作用,选择合适的淋洗溶液不仅可以淋洗去除分子印迹聚合物由于非特异性作用力结合的杂质,同时能最大程度地保留目标化合物在小柱中,实现目标化合物与干扰物的分离,从而提高检测的灵敏度和准确性。因此,本文考察了二氯甲烷及乙腈-二氯甲烷(1∶99,2∶98,3∶97,4∶96,v/v)混合溶液对分子印迹固相萃取小柱的淋洗情况。如图7a所示,NS-MISPE及NISPE对NS的回收率都随着乙腈含量的增加而下降,当淋洗溶剂为二氯甲烷时,回收率最高,因此本文选择二氯甲烷作为淋洗溶剂。同时对淋洗液的体积进行了优化。由图7b可知,当淋洗液的体积为4 mL时,从NS-MISPE和NISPE上淋洗下来的NS含量差异最大,而NS-MIPMs对NS的作用为特异性吸附,NISPE为非特异性吸附,这说明当使用4 mL二氯甲烷作为NS-MISPE的淋洗液时,可以最大程度地将非特异性吸附的杂质从小柱中分离,因此本文最终选择4 mL的二氯甲烷作为淋洗液。
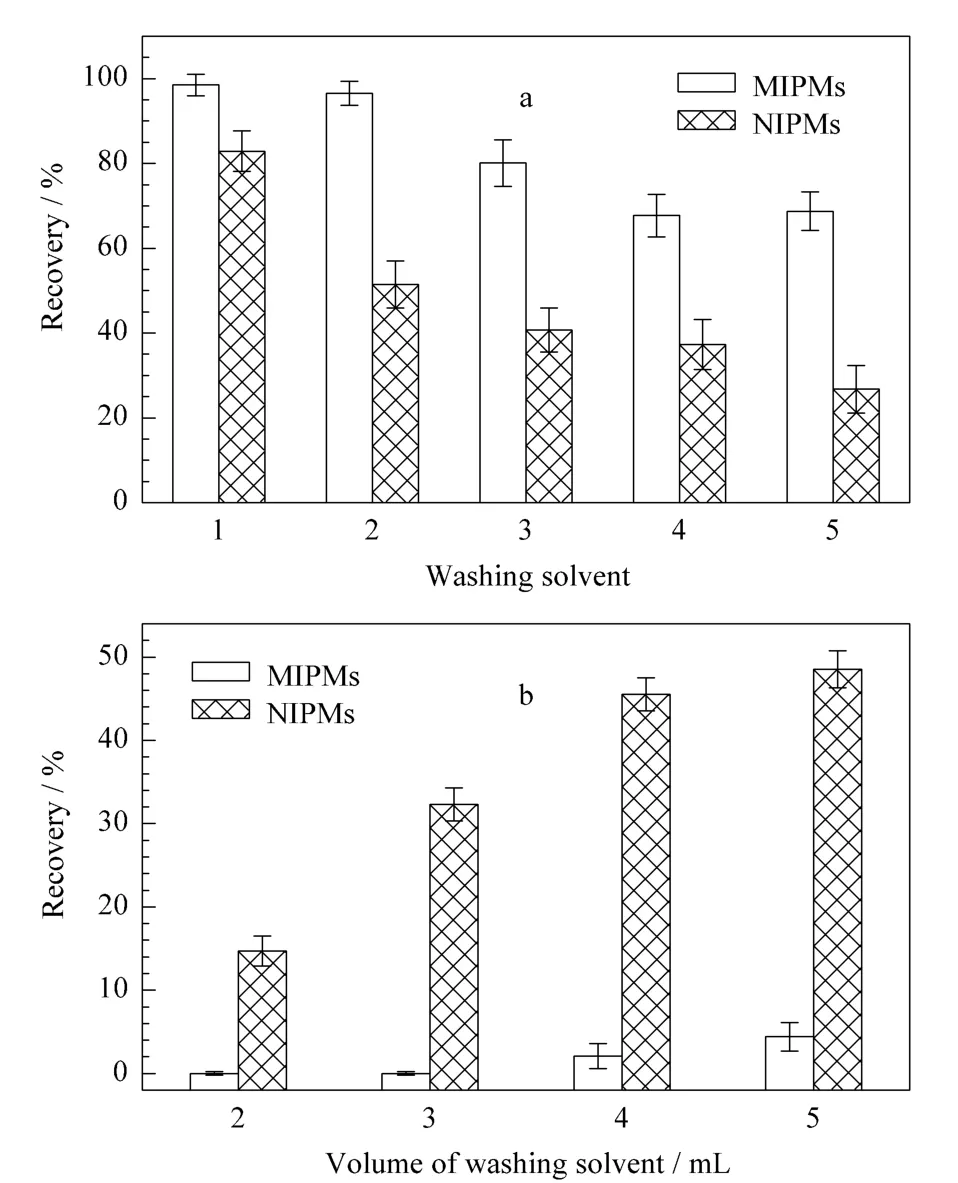
图7 NS-MISPE淋洗溶剂的(a)种类和(b)体积的影响Fig.7 Influences of(a)washing solvent and(b)washing solvent volume for NS-MISPE
对于洗脱溶液的选择,要能够完全地将吸附在分子印迹固相萃取小柱中的目标化合物洗脱下来,本文选择3 mL的二氯甲烷/乙腈(2∶1,v/v)混合溶液作为洗脱液,回收率可达98%。
2.6.2 NS-MISPE-HPLC检测土壤中烟嘧磺隆残留的方法
准确配制一系列质量浓度为0.01、0.02、0.05、0.1、0.2、0.5、1、2 mg/L 的烟嘧磺隆标准溶液,按1.2节条件分别进行测定,以分析物的峰面积y对质量浓度x(mg/L)做标准曲线,得到y=33782x-1898.7,r2=0.9986,表明烟嘧磺隆在 0.01~1 mg/L范围内具有良好的线性关系。按信噪比(S/N)=3计方法的检出限(LOD)为0.002 mg/kg,说明该方法适用于土壤中烟嘧磺隆的残留检测。
同时对土壤进行添加试验,考察NS-MISPEHPLC方法的准确度和精密度。分别在10.00 g土壤样品中添加0.02、0.2、1 mg/kg 3个水平的烟嘧磺隆标准样品,按1.2节和1.9节所述方法处理样品并进样分析,平行测定5次,得到加标回收率和相对标准偏差分别为85.1%和1.9%、82.2%和4.3%、86.3%和2.4%,说明烟嘧磺隆的回收效果较好,基本能够满足农药残留分析方法的性能要求。经过NS-MISPE净化前后的样品的色谱图见图8。
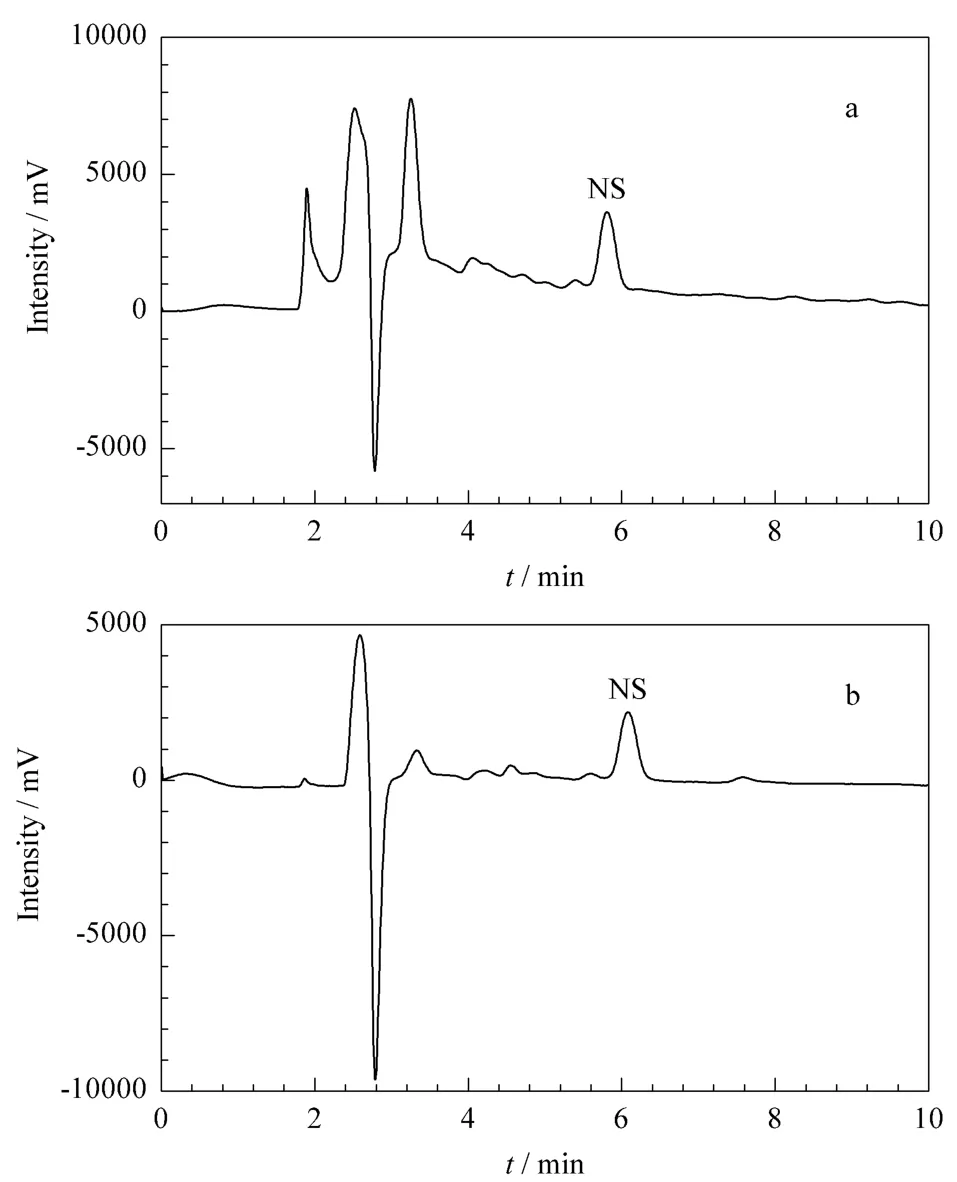
图8 加标土壤样品的色谱图Fig.8 Chromatograms of a spiked soil sample
3 结论
采用沉淀聚合的方法,在氯仿中合成了烟嘧磺隆分子印迹聚合物微球,并优化了聚合工艺。该工艺制备的聚合物不需要研磨和过筛,并具有比传统分子印迹聚合物更高的选择性和吸附性能。通过紫外分光光度计考察了NS与MAA的作用机理为一个MAA和一个NS以氢键相结合。吸附动力学实验表明NS-MIPMs对模板分子NS具有较高的吸附容量和较快的吸附速度。将NS-MIPMs应用到固相萃取技术中,建立了NS-MISPE-HPLC检测土壤中烟嘧磺隆残留的方法,回收效果较好,基本能够满足农药残留分析方法的性能要求。
[1]Sarmah A K,Sabadie J.J Agric Food Chem,2002,50(22):6253
[2]Seccia S,Albrizio S,Fidente P,et al.J Chromatogr A,2011,1218(9):1253
[3]Ayano E,Kanazawa H,Ando M,et al.Anal Chim Acta,2004,507(2):211
[4]Zhou Q X,Liu J F,Cai Y Q,et al.Microchem J,2003,74(2):157
[5]Fenoll J,Hellin P,Flores P,et al.Chemosphere,2012,87(8):954
[6]Eri A,Hideko K,Masanori A,et al.Anal Chim Acta,2004,507(2):211
[7]Li J H,Wen Y Y,Chen L X.Chinese Journal of Chromatography(李金花,温莹莹,陈令新.色谱),2013,31(3):181
[8]Wulff G,Sarhan A,Zabrocki K,et al.Tetrahedron Lett,1973,44(14):4329
[9]Lasáková M,Jandera P.J Sep Sci,2009,32(5/6):799
[10]Li Z M.[MS Thesis].Xiangtan:Xiangtan University(李兆敏.[硕士学位论文].湘潭:湘潭大学),2010
[11]She Y X,Cao W Q,Shi X M,et al.J Chromatogr B,2010,878(23):2047
[12]Tang K J,Chen S W,Gu X H,et al.Anal Chim Acta,2008,614(1):112
[13]Geng L Y,Kou X,Lei J D,et al.J Chem Technol Biotechnol,2012,87(5):635
[14]Yang W H,Yan S L,Wei C,et al.Acta Polymerica Sinica(杨卫海,严守雷,卫晨,等.高分子学报),2010(10):1163
[15]Lai J P,Niessner R,Knopp D,et al.Anal Chim Acta,2004,522(2):137
[16]Yan S L,Gao Z X,Fang Y J.Acta Polymerica Sinica(严守雷,高志贤,房彦军.高分子学报),2006(1):160
[17]Dai C M,Zhou X F,Zhang Y L,et al.J Hazard Mater,2011,198:175
[18]Chen A Z,Dong X C.Chemistry(陈安珍,董襄朝.化学通报),2006,69(1):1
[19]Jiang Y,Kim D.Chem Eng J,2011,166(1):435
[20]Qiao F X,Geng Y R,He C Q,et al.J Chromatogr B,2011,879(27):2891
