基于HLA等位基因频率的全球人群亲缘关系初探
2014-09-12黄庆海吴亚萌李震谢维
黄庆海,吴亚萌,李震,谢维
(“发育与疾病相关基因”教育部重点实验室 东南大学生命科学研究院,江苏 南京 210096)
编码人类白细胞抗原(human leukocyte antigen,HLA)的基因是人体最为复杂、多态性最高的遗传系统,属双亲共显性遗传,也是最早用于研究人群遗传亲缘关系的标记物之一[1]。20世纪70年代初期,HLA血清型(低分辨率)即已被用于人群亲缘关系研究,但由于HLA血清型属表现型,分辨率低,不能完全反映群体遗传关系。20世纪90年代以来,随着HLA高分辨率分型技术中的相继应用,HLA基因型(高分辨率)分型技术越来越成熟,多样性也越来越多。国际组织相容性工作组(The International Histocompatibility Working Group, IHWG)在第11届[2]、12届[3]、13届[4]学术会议中专题报道了大量人群HLA基因型的分布特征,13届(2001年)学术会议结束后,专门设立了人类学/人类遗传多样性专项,继续采集人群HLA及其免疫分子的多样性数据。再加上很多学者关注与HLA群体分布特征有关的骨髓捐献库分型、HLA与疾病相关性等研究,HLA基因型数据越来越多,可使用的分析数据也越来越丰富。尽管这些数据为人类学研究,特别是人口历史、人群扩张途径等提供了很好的资源,但却未见利用如此大量的数据勾画出全球人群间的亲缘关系。有观点认为,HLA并不适合用于历史基因组的研究[5]。我们收集了全球110个人群的HLA高分辨率配型数据,对所有数据的等位基因命名和统计方法进行了归一化处理,运用N- J法构建了多种全球人群关系树,证明HLA分子的多态性无论是在全球范围还是在某一局部地区都能很好地用于分析人群的亲缘关系。
1 资料与方法
1.1 数据收集与处理
本研究收集了PubMed (www.ncbi.nlm.nih.gov/pubmed)、CNKI (China National Knowledge Infrastru-cture, www.cnki.net)和IHWG会议论文集[2- 4]中发表的、针对本地(非近期迁徙而来)不相关健康人群的高分辨率(配型在2节,旧命名系统的4位及以上)的HLA分型数据的文献,并且去除那些相似作者群体研究相同地区、相同民族人群的文献。将这些人群数据构建成本地VFP(Visual FoxPro, VFP)数据库,后续所有的计算和编程都在VFP环境中执行。本研究根据国际人类学研究惯例[6- 8],将全球人群分为10个区域:撒哈拉沙漠以南非洲(sub- Saharan Africa, SSA)、北非(North Africa, NAF)、欧洲(Europe, EUR)、 西南亚(Southwest Asia, SWA)、 东北亚(Northeast Asia, NEA)、东南亚(Southeast Asia, SEA)、大洋洲(Oceania, OCE)、 澳大利亚(Australia, AUS)、北美洲(North America, NAM)和南美洲(South America, SAM)。
考虑到国际HLA命名系统在不断更新,为方便数据间进行相互比较,本研究将所有人群数据的等位基因均更新为最新的名称(HLA ambiguity release version 3.13.0, www.ebi.ac.uk/ipd/imgt/hla/ambig.html[9]),例如将HLA- A*010101替换为HLA- A*01∶01∶01。同样为了便于数据比较,我们将HLA的分型精度定位为2节,将编码抗原肽结合凹槽的HLA Ⅰ类基因的第2、第3外显子相同的等位基因视为同一种等位基因,例如将HLA- A*01∶01∶01∶01, A*01∶01∶01, A*01∶01∶02, A*01∶04N, A*01∶22N等归一为HLA- A*01∶01;将编码抗原肽结合凹槽的HLA Ⅱ类基因的第3外显子相同的等位基因视为同一等位基因,例如HLA- DRB1*01∶01包括HLA- DRB1*01∶01∶01, DRB1*01∶01∶02, DRB1*01∶50等。
为了提高数据小数的精度,以达到更为精确的比较效果,所有人群的等位基因频率用公式1和公式2重新进行计算:
n=Af’*2N
(1)
Af=n/2N
(2)
其中的n代表等位基因频数,Af’代表原文中的等位基因频率,Af代表重新计算出的等位基因频率(下同),N代表样本个体数量。
对于那些用表现型频率统计的文献,根据Hardy- Weinberg平衡原理,用公式3将表现型频率转换为基因型频率:
(3)
其中的Pf代表表型频率。为达到更为稳定的计算效果,本研究通过公式4将同一人群的HLA- A,- B, 和- DRB1座位的等位基因频率合并成一个虚拟的、联合的HLA- ABD座位的等位基因频率:
AfABD=(AfA+AfB+AfDRB1)/3
(4)
其中AfABD代表联合座位HLA- ABD的等位基因频率,AfA、AfB和AfDRB1分别代表HLA- A,- B和- DRB1座位的等位基因频率。
1.2 构建人群关系树
本研究选择马氏距离(亦称为曼哈顿距离,Manhattan distance)[10- 11]计算人群间的遗传距离,距离计算如公式5所示:
(5)
其中xi和yi分别代表第x个人群和第y个人群中的第i个等位基因频率,M为参与比较的两个人群共同拥有的等位基因数目。再选择N- J法(neighbor- joining method)[12],用MEGA5 (http://www.megasoftware.net/)[13]构建人群关系树。
本研究中不同地区的人群数量差异较大,最多的是东南亚地区,人群数量为33个,最少的是澳大利亚地区,人群数量仅为2个。为了分析不同地区的人群数量不同是否影响人群关系树的拓扑结构,我们在每个地区中随机挑选了2个人群,构建了10个地区20个人群的关系树。为了更进一步分析这种影响,更好地区分不同地区人群的亲缘关系,本研究计算了各地区人群间的平均遗传距离,再用平均遗传距离重新构建了10个地区人群间的关系树。各地区人群间的平均遗传距离如公式6所示:
(6)
其中M代表参与比较的某一地区人群的数量,N代表参与比较的另一地区人群的数量。Di,j代表参与比较的第一个地区的第i个人群与第二个地区的第j个人群间的遗传距离。
为了便于与以往其他学者运用mtDNA、NRY、全基因组SNP或者其他分子标记物对各地人群研究的结果进行比较,作者以取自津巴布韦的绍纳人(Shona)作为人群关系树的根节点,根据人群的地理位置对一些人群在关系树的分布进行了相应旋转。
2 结果与分析
本研究收集了国内、外用于人类学、民族学、骨髓库配型以及疾病对照研究产生的HLA高分辨率配型数据,总计110个健康本地人群,分布于非洲、欧洲、亚洲、北美洲、南美洲以及大洋洲(太平洋岛屿和澳大利亚)。HLA- A、- B、- DRB1座位的样本量分别为454 071、455 233和496 221个染色体,合并成联合的HLA- ABD座位后代表1405 525个染色体。HLA- ABD由HLA- A、- B和- DRB1 三个座位合并而来,其携带了更为丰富、更为稳定的遗传信息,由此而构建的人群关系也更为稳定,更具代表性。因此,我们的结果与讨论将主要针对HLA- ABD座位展开。
用联合的HLA- ABD构建的全球110个人群的关系树显示,各大区域内的人群相互聚类,不同区域间的人群完全分离(图1)。撒哈拉以南非洲人群聚于人群树的根部,这与本研究选择了津巴布韦人作为关系树的根节点有关。美籍非洲人居于南部非洲人群的中部,喀麦隆人、南非共和国人更靠近关系树的根部,西部非洲人群则与北非人群靠近。北非的3个人群与西亚人群聚为一大支,显示出北非的阿拉伯人或者柏柏尔人(Berbers)与西亚的阿拉伯人在遗传上有较高的相似性。欧洲人群将西南亚人群分为两部分,一部分是向北非人群靠近的西亚人群,另一部分是与东亚人群靠近的南亚次大陆人群(印度人);南欧人群与中/北/西欧人群在法国相汇,暗示了欧洲南北部人群的基因成分是沿地中海东西两岸进行交流的,这也与用全基因组的SNP分析欧洲人群的遗传结构的结果[14]相一致。北亚人群(本分析中主要指韩国人和日本人,也包括部分中国北方少数民族)尽管与东南亚人群聚为一大支,但更接近于中亚人群,暗示了东北亚人群与中亚人群有较多的基因交流。14个汉族人群聚为一支,并基本以由北向南的顺序相邻接,南方汉族人群与北方汉族人群在长江一带相汇,该结果与早期的多个基因座位联合研究的结果[15]相一致,再一次说明中国南北方人群的分界线是长江天堑,而不是作为中国南北方气候分界线的秦岭淮河一线。中国台湾原住民(高山族和平埔族)与菲律宾人、巴布亚新几内亚人等聚类,该支与澳大利亚原住民比较靠近,显示了作为南岛语系的台湾原住民的遗传特征,以及与南太平洋岛屿人群的遗传相似性。美洲印第安人群聚为一大支,该支又分为两簇,一个是北美印第安人,另一支是南美印第安人,并且基本按由北向南的顺序排列。
为了测试不同地区的人群数量不同是否影响人群关系树的拓扑结构,我们从各地理区域内随机挑选 2个共计20个人群构建了新的人群树。结果显示,各区域内的两个人群聚为一支,各区域之间人群的毗邻关系与前述110个人群的关系树的总体拓扑结构一致(图2),说明不同地区人群数量的不平衡并不影响人群树的总体拓扑结构。为了更进一步验证这一结果,本研究用各地区间人群的平均遗传距离构建了10个区域的关系树,仍然显示出与前述两种关系树相一致的拓扑结构,自人群关系树的根部至顶部,分别是撒哈拉以南的非洲、北非、欧洲、西南亚、东北亚、东南亚、大洋洲、澳洲、北美洲和南美洲(图3)。
3种方法构建的人群树都能稳定展示出全球人群同样的亲缘关系,我们也用HLA- A、- B、- DRB1三个座位的等位基因频率分别构建了全球人群的关系树,并且又用Cavalli- Sforza距离、修改后的Cavalli- Sforza距离、Nei的标准距离和Rogers- Wright距离等常用的另外4种遗传距离算法[11]构建了基于HLA- ABD等位基因频率的全球人群关系树。不同数据、不同算法构建的人群树均显示了同样的结果(数据未发表),并且与应用全基因组SNP构建的全球人群关系树有着相似的拓扑结构[16- 17]。说明作为常染色体1个片段的HLA基因能够稳定地显示全球人群的亲缘关系。
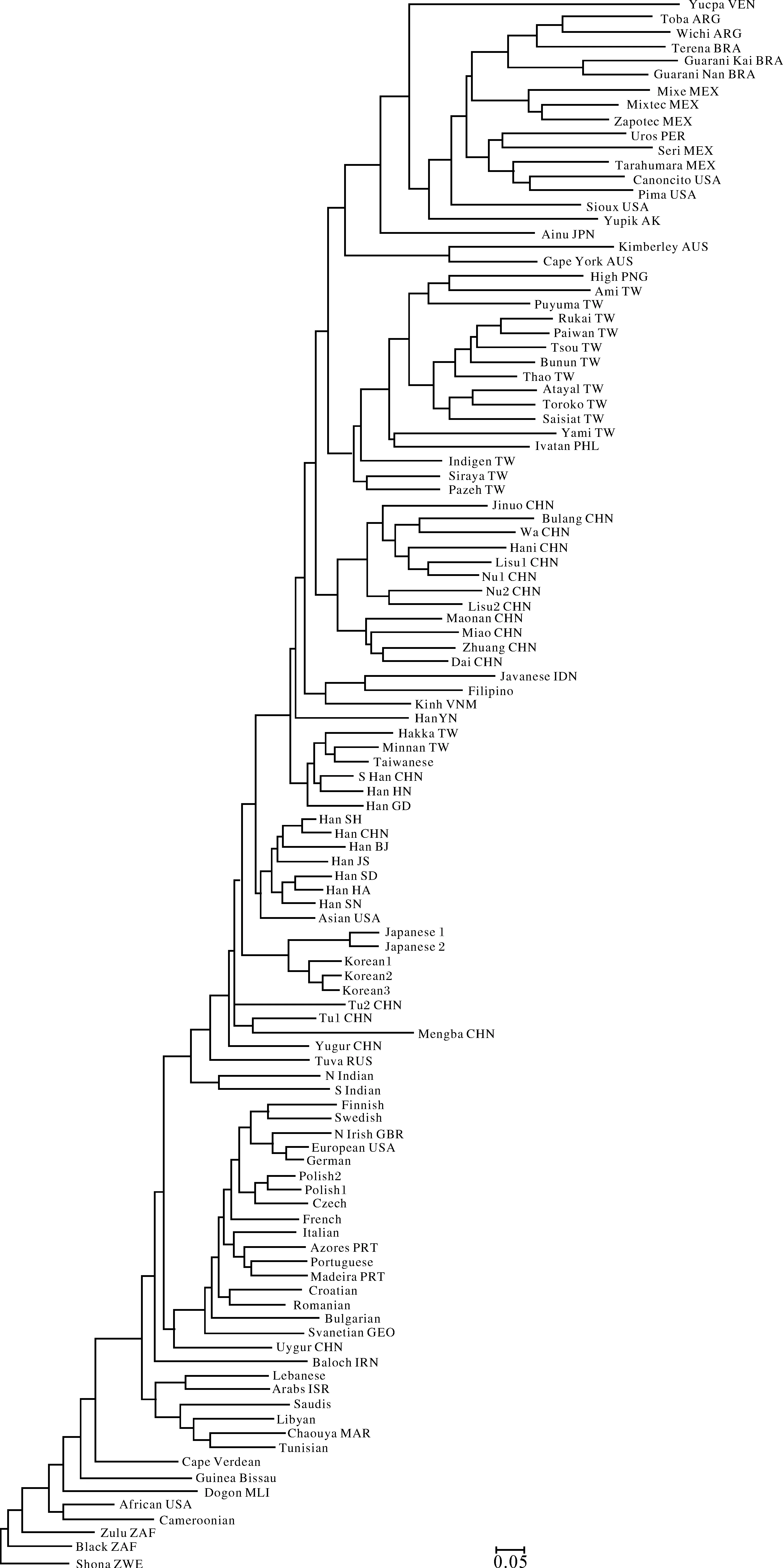
图1用HLA-ABD等位基因频率构建的10地区110个人群的N-J树
Fig1N-JTreeof110populationsfromthetengeographicbasedonallelefrequencyofcombinedHLA-ABD
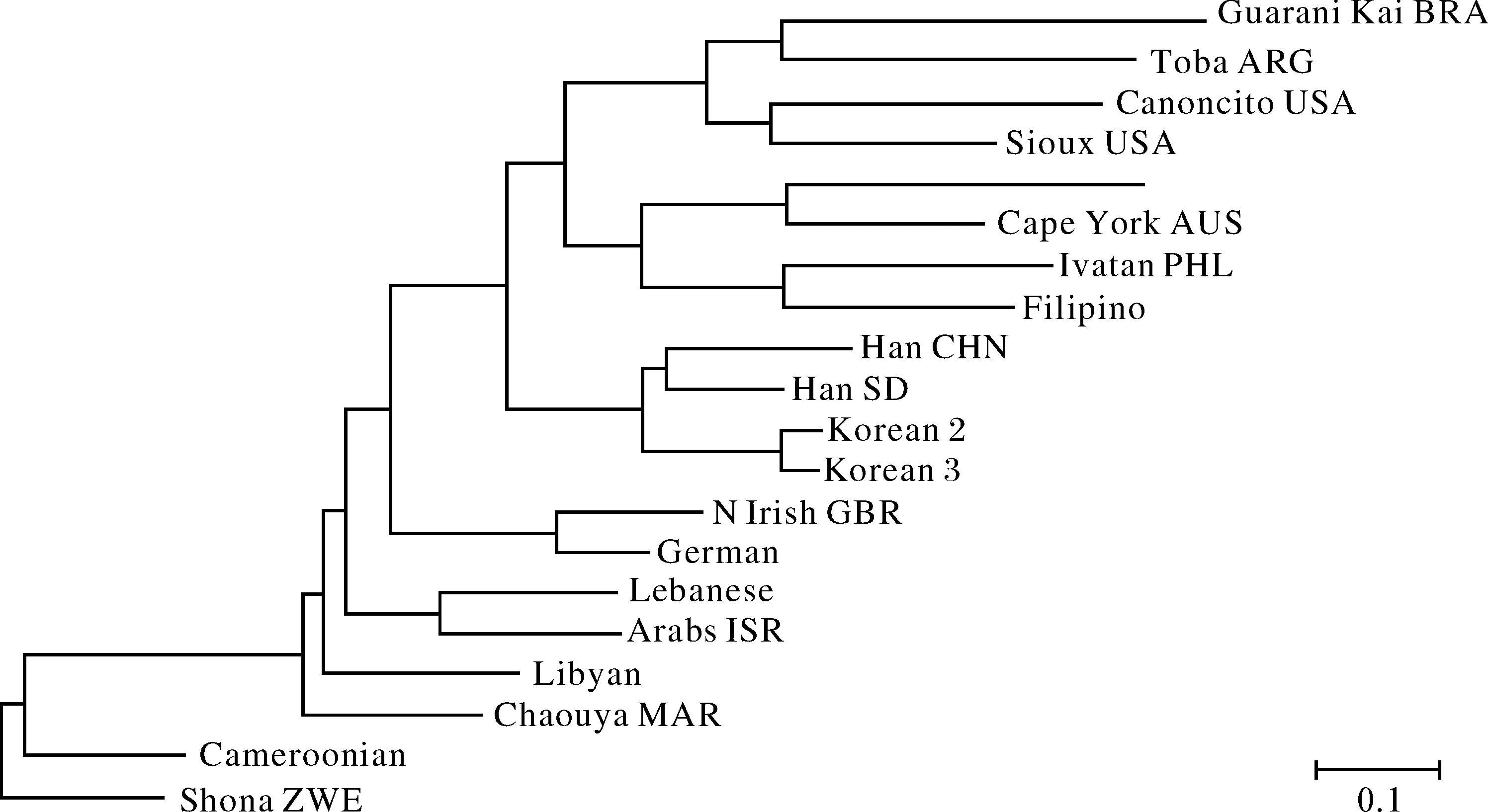
图2用HLA-ABD等位基因频率构建的10地区20个随机人群的N-J树
Fig2N-JTreeof20populationsselectedrandomlyfromeachofthetengeographicregionsbasedonallelefrequencyofcombinedHLA-ABD
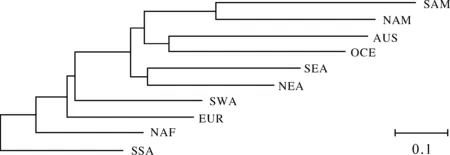
图3用各地区间平均遗传距离构建的10地区人群的N-J树
Fig3N-JTreeofpopulationsfromthetengeographicpopulationgroupsusingtheaveragedistancesbetweenanytwogeographicregions
3 讨 论
20多年来,如此大量的HLA配型数据为人们提供了很好的分析人群亲缘关系的资源,但却未见应用该基因多态性构建全球人群关系树的报道。一般归结于HLA与环境的选择压力、遗传性或者病原性疾病的选择压力有关。在本研究中,美籍非洲人已经在北美洲定居了300多年,但在人群关系树中仍与非洲的喀麦隆人聚类,美籍欧洲人、美籍亚洲人也都与其迁出地的人群聚类,至少说明在短期内,环境的压力并不会对人群的HLA频率分布特征产生影响,人群的HLA频率分布特征是人群固有的遗传特征。本研究也将一些病人的HLA等位基因频率与正常健康人群的进行了比较对照,结果各地的病人群体仍与各地的健康人聚类,而并非不同地区的同一类病人群体聚类(数据未发表)。某一类病人在总人群中只占一小部分,并且那些与疾病相关的某个座位的某一个或某几个等位基因频率的改变对人群的总体HLA频率分布特征影响很小。本研究应用HLA等位基因频率构建的人群关系树与用中性无选择压力的分子标记物—全基因组SNP构建的关系树有相似的拓扑结构[16- 17]和人群亲缘关系,同样证明HLA基因的多态性是各人群固有的遗传特征,适合作为分子标记物用于全球人群的亲缘关系分析。
我们在数据整理和分析过程中发现,那些混合人群会对关系树的结构产生较大影响,例如,很多研究发现瑞典的少数民族Sami[18- 20]存在亚洲基因成分,当我们将Sami的数据并入研究时发现,Sami与芬兰人、瑞典的非Sami人聚为一小支,该支脱离欧洲主支而向亚洲方向靠近,并且东北亚人群在关系树中的位置也发生了变化,从而影响了对芬兰人、瑞典的主要人群和东北亚人群的遗传关系分析。表明这些混合人群将吸引他的几种祖先人群相互靠近,从而打乱了关系树的拓扑结构,影响了对主要人群的遗传分析。除了瑞典的Sami外,我们在研究过程也排除了阿留申群岛人(excluded Aleuts)[21]、新西兰的毛里人(Maori)[22]和复活节岛人(Easter Islander)[23]的配型数据,因为原文明确表明取样人群存在与近期迁入人群的混血成分。这种现象提醒我们,在进行全球人群的数据分析时,应当非常仔细地甄别那些近期混合人群,注意这些人群对关系树造成的影响。
20多年来,随着HLA高分辨率配型方法的不断改进,统计方法也各不相同,特别是在用限制片段长度多态性(restriction fragment length polymorphism, RFLP)配型时,有的文献使用的是基因型频率统计,有的文献使用的是表现型频率统计,这两种统计方法的基本原理完全不同,需要通过Hardy- Weinberg平衡原理将表型频率转换成基因型频率后才能进行比较。另外,不同的文献HLA配型的分辨率也不相同,需要对其归一化后才能进行统一比较。本研究以编码PBR区域的基因多态性为基准,将配型数据定为2节,例如,将HLA*02∶01∶01,HLA*02∶01∶02等等位基因的频率并为HLA*02∶01的频率统计。经过数据重新处理和统计后,长达20多年的配型数据才能进行相互比较,这也是多年来未能将如此丰富的人类学数据用于人类学研究和分析的主要原因。
经过对等位基因名称、统计方法等的标准化和归一化,出自不同实验室、不同实验方法、不同统计方法的HLA等位基因频率数据可以进行相互比较,并且构建了完整清晰的人群关系树,无论是在全球范围内还是在各个地理区域,根据关系树都能很好地解释和分析人群间的遗传差异性和相似性,说明HLA基因的多态性也是一种高效的研究人类学的分子标记物。
致谢上海交通大学医学院范丽安教授和刘祥箴同学对本研究给予了帮助。
[1] GAUDIERI S,DAWKINS R L,HABARA K,et al.SNP profile within the human major histocompatibility complex reveals an extreme and interrupted level of nucleotide diversity[J].Genome Res,2000,10(10):1579- 1586.
[2] IMANISHI I,AKAZA T,KIMURA A,et al.Allele and haplotype frequencies for HLA and complement loci in various ethnic groups[C]// TSUJI K,AIZAVA M,SASAZUKI T.HLA 1991:Proceedings of the Eleventh International Histocompatibility Workshop and Conference,vol I.Oxford,UK:Oxford University Press,1991:1065- 1220.
[3] BODMER J,CAMBON- THOMSEN A,HORS J,et al.Report of the anthropology component[C]// CHARRON D,FAUCHET R.HLA:Proceedings of the Twelfth International Histocompatibility Workshop and Conference,Vol I.Paris,France:EDK,1997:269- 274.
[4] MACK S,ERLICH H.Anthropology/Human genetic diversity joint report[C]// HANSEN J.Immunobiology of the human MHC:Proceedings of the 13th International Histocompatibility Workshop and Conference,Vol I.Seattle,USA:IHWG Press,2007:557- 766.
[5] ARNAIZ- VILLENA A.Historic genomics:an emergent discipline[J].Hum Immunol,2001,62(9):869- 870.
[6] SOLBERG O,MACK S,LANCASTER A,et al.Balancing selection and heterogeneity across the classical human leukocyte antigen loci:a meta- analytic review of 497 population studies[J].Hum Immunol,2008,69(7):443- 464.
[7] MEYER D,SINGLE R,MACK S,et al.Signatures of demographic history and natural selection in the human major histocompatibility complex loci[J].Genetics,2006,173(4):2121- 2142.
[8] MACK S,SANCHEZ- MAZAS A,MAYER D,et al.Methods used in the generation and preparation of data for analysis in the 13th International Histocompatibility Workshop[C]// HANSEN J .Immunobiology of the human MHC:Proceedings of the 13th International Histocompatibility Workshop and Conference,Vol I.Seattle,USA:IHWG Press,2007:564- 579.
[9] ROBINSON J,HALLIWELL J,McWILLIAM H,et al.The IMGT/HLA database[J].Nucleic Acids Res ,2013,41(Database issue):D1222- 1227.
[10] POWELL J,LEVENE H,DOBZHANSKY T.Chromosomal polymorphism in drosophila pseudoobscura used for diagnosis of geographic origin[J].Evolution,1972,26(4):553- 559.
[11] NEI M,TAJIMA F,TATENO Y.Accuracy of estimated phylogenetic trees from molecular data.Ⅱ.Gene frequency data[J].J Mol Evol,1983,19(2):153- 170.
[12] SAITOU N,NEI M.The neighbor- joining method:a new method for reconstructing phylogenetic trees[J].Mol Biol Evol ,1987,4(4):406- 425.
[13] TAMURA K,PETERSON D,PETERSON N,et al.MEGA5:molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance,and maximum parsimony methods[J].Mol Biol Evol,2011,28(10):2731- 2739.
[14] SELDIN M,SHIGETA R,VILLOSLADA P,et al.European population substructure:clustering of northern and southern populations[J].PLoS Genet,2006,12:e143.
[15] 杜若甫,肖春杰,CAVALLI- SFORZA L.用38个基因座的基因频率计算中国人群间遗传距离[J].中国科学:C辑,1998,28(1):83- 89.
[16] LI J,ABSHER D,TANG H,et al.Worldwide human relationships inferred from genome- wide patterns of variation[J].Science,2008,319(5866):1100- 1104.
[17] ABDULLA M,AHMED I,ASSAWAMAKIN A,et al.Mapping human genetic diversity in Asia[J].Science,2009,326(5959):1541- 1545.
[18] TAMBETS K,ROOTSI S,KIVISILD T,et al.The western and eastern roots of the Saami- - the story of genetic "outliers" told by mitochondrial DNA and Y chromosomes[J].Am J Hum Genet,2004,74(4):661- 682.
[19] INGMAN M,GYLLENSTEN U.A recent genetic link between Sami and the Volga- Ural region of Russia[J].Eur J Hum Genet ,2007,15(1):115- 120.
[20] JOHANSSON A,INGMAN M,MACK S,et al.Genetic origin of the Swedish Sami inferred from HLA class Ⅰ and class Ⅱ allele frequencies[J].Eur J Hum Genet,2008,16(11):1341- 1349.
[21] MOSCOSO J,CRAWFORD M,VICARIO J,et a.HLA genes of Aleutian Islanders living between Alaska(USA)and Kamchatka (Russia) suggest a possible southern Siberia origin[J].Mol Immunol,2008,45(4):1018- 1026.
[22] TRACEY M,CARTER J.Class Ⅱ HLA allele polymorphism:DRB1,DQB1 and DPB1 alleles and haplotypes in the New Zealand Maori population[J].Tissue Antigens,2006,68(4):297- 302.
[23] THORSBY E,FLAM S,WOLDSETH B,et al.Further evidence of an Amerindian contribution to the polynesian gene pool on Easter Island[J].Tissue Antigens,2009,73(6):582- 585.
