组蛋白乙酰化与缺血性心肌病
2014-09-12卢圣锋何苏云朱冰梅
黄 艳 卢圣锋 何苏云 朱冰梅
(南京中医药大学第二临床医学院,江苏 南京 210023)
缺血性心肌病(ICM)是指由于长期心肌缺血导致心肌局限性或弥漫性纤维化,从而产生心脏收缩和(或)舒张功能受损,引起心脏扩大或僵硬、充血性心力衰竭、心律失常等一系列临床表现的综合征。随着全国人口老龄化程度增加, ICM发病率、死亡率急剧上升;在美国约500万心力衰竭患者中,至少有350万左心室收缩、舒张功能不全者系ICM所致,已经造成严重的社会经济负担〔1,2〕。因其机制尚未完全明确,缺乏特异性的治疗手段,一直是医学研究的热点。表观遗传学(Epigenetics)是研究基因核苷酸序列不发生改变的情况下,基因表达了可遗传的变化的一门遗传学分支学科;认为遗传信息主要以DNA甲基化、组蛋白修饰等方式保存,可逆性调节真核基因的表达〔3〕。越来越多的证据表明,ICM是一种与遗传、环境因素等相关的疾病。组蛋白乙酰化在ICM中的作用越来越受到重视,通过组蛋白乙酰化酶和去乙酰化酶相互作用,在缺血缺氧所致心肌细胞肥大、间质纤维组织增生、广泛的心肌纤维化中扮演重要角色,这将为ICM的治疗提供新的治疗靶点,为ICM研究提供新的思路。
1 组蛋白乙酰化修饰调控基因转录
1.1组蛋白乙酰化修饰的生物特性 组蛋白乙酰化修饰是基于赖氨酸(Lys)残基N末端的乙酰化修饰,是目前研究最多最广泛的组蛋白修饰形式。组蛋白乙酰化和去乙酰化能改变核小体周围的电荷及染色质构型,当染色质处于相对松弛的状态,暴露某些转录因子的识别位点和结合平台,利于转录因子与DNA结合〔1~4〕。组蛋白末端的共价修饰还构成了独特的组蛋白密码,组蛋白的多种修饰方式相互协同作用,构成基因特异性表达的重要基础。目前研究组蛋白乙酰化修饰的位点较多,主要发生在H3、H4上,不同的乙酰化修饰位点均为激活基因转录的作用,组蛋白乙酰化、甲基化修饰共同作用参与转录的激活与沉默〔5〕(见表1)。

表1 组蛋白修饰和相关的转录调控作用
1.2组蛋白乙酰化修饰相关酶类及相互作用 组蛋白乙酰基转移酶(HAT)催化组蛋白和非组蛋白乙酰化,是最早被发现的与转录有关的组蛋白修饰;而组蛋白去乙酰化酶(HDAC)催化组蛋白去乙酰化。HAT和HDAC共同调节组蛋白乙酰化水平,是组蛋白修饰最重要的方式,也是调控基因表达最主要的驱动力〔6〕,它们各自形成的基因转录调控复合物分别称为辅助激活因子(CoA)和辅助抑制因子(CoR)。前者可使DNA打开,组蛋白乙酰化,激活基因转录;后者可使DNA关闭,组蛋白去乙酰化,抑制基因转录,两者共同作用实现基因组整体水平的调控(见图1〔7〕)。
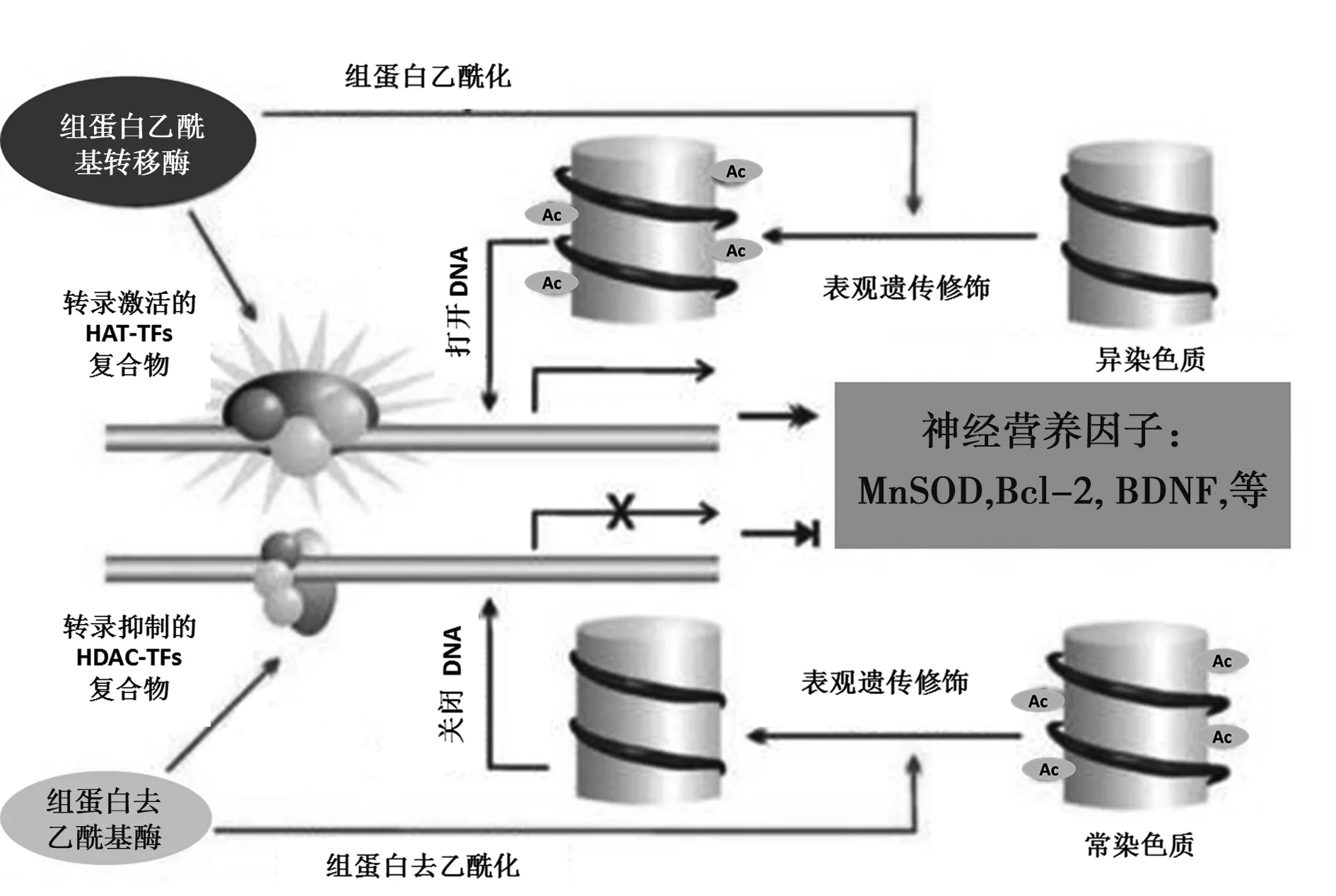
乙酰化转录因子活化的结构域招募HATs复合物,打开染色质,催化附近的组蛋白乙酰化,促进基因激活;相反,去乙酰化转录因子抑制的结构域招募 HDACs复合物,关闭染色质,催化附近组蛋白去乙酰化,转录抑制
HAT的主要功能是将乙酰辅酶A的乙酰基转移到组蛋白的赖氨酸残基上,可使核小体组蛋白乙酰化,也可使非组蛋白乙酰化。目前研究发现,20多种HATs已被鉴定具有内源性的A型HATs活性〔8〕(见表2)。
HDAC是一类催化组蛋白的赖氨酸上去乙酰基的酶,在染色质固缩和基因调控上起着关键性的作用。目前已知的HDACs按其同源性分为4类(见表3)。
Ⅰ类和Ⅱ类HDACs的活性可以被共同的抑制剂如曲古抑菌素A和n-丁酸等所抑制;Ⅲ类HDACs对这些抑制剂不敏感,但其活性可以被烟酰胺(NAM )抑制〔13,14〕。

表2 HATs的分类和主要作用

表3 HDACs分类及主要作用
2 组蛋白乙酰化修饰与ICM
ICM的病理变化发展过程中,长期慢性缺血缺氧导致心肌细胞能量生成障碍,心肌细胞内Ca2+超载等病理生理改变引发心肌细胞肥大、凋亡、胚胎基因再表达、胞外基质堆积及其组成等变化,导致心肌肥厚、心室重构和心脏的舒缩功能障碍。大量研究表明,组蛋白乙酰化在心肌细胞肥大、心衰等心血管疾病过程中扮演重要角色,直接参与ICM的发生、发展过程〔15,16〕。
2.1组蛋白乙酰化与肌供氧需氧平衡失调 ICM患者,尤其是充血型ICM,往往伴有多支冠状动脉发生显著性粥样硬化性狭窄,导致长期的心肌供氧和需氧之间不平衡,随着病程进展出现心肌细胞肥大、凋亡。最近的研究表明,HDACs抑制剂能抑制缺氧导致的胚胎基因(GATA)4再表达,减少供需氧失衡导致的血管和心肌损伤,从而保护缺血心肌细胞的功能,提示组蛋白乙酰化在控制心脏的基因表达中具有重要作用〔17〕。此外,HDAC2有可能参与缺氧导致心肌细胞肥大的机制,而HDACs抑制剂可抑制心肌细胞肥大,减少对心脏的缺血性损伤及其梗死面积,减少心功能受损,延迟心衰的发作时间,在心脏重构中发挥重要调节作用〔18,19〕。微小RNA126是HAT/HDAC的缺氧诱导目标,在心肌细胞中的活性可通过激活细胞和促进血管生成途径对缺血缺氧心肌产生保护作用〔20〕。组蛋白乙酰化参与胚胎基因再表达的调节,抑制心肌细胞肥大,还参与缺血缺氧心肌细胞的激活、心脏血管的生成。
2.2组蛋白乙酰化与心肌细胞能量代谢障碍 ICM病程中伴有心肌细胞能量代谢障碍,心肌缺血可激活交感神经,使心率加快,心肌耗氧量增加,影响肌浆网钙ATP酶的活性,Ca2+转入肌浆网内,加速心肌细胞凋亡,进而减弱心肌收缩力。当心肌缺血缺氧时,糖酵解变为能量的主要来源,进而引起心肌能量代谢障碍。在心肌细胞中,叉形头转录因子(FOXO3A)、葡萄糖转运蛋白(GLUT)4在心肌细胞中参与糖代谢,心肌缺血缺氧引起GLUT4转位至细胞膜上,引起能量代谢失衡。SIRT1属于Ⅲ类HDACs,在缺血心肌能量代谢中参与调节内分泌信号,调节FOXO3A、GLUT4,同时调节脂代谢相关的过氧化物酶体增殖物激活受体(PPAR)γ、PPARγ共激活因子PGC-1α,进而调节心肌细胞的能量代谢平衡,在缺血心肌细胞能量代谢中发挥重要作用〔21〕。
2.3组蛋白乙酰化与缺血对心功能和心肌电活动的影响 心肌缺血导致部分心肌细胞坏死,心排血量和每搏量减少,左心室舒张末期容量增加、长期缺血缺氧可致左心室收缩期容量也增加、心脏扩大和心力衰竭。有研究发现,细胞因子的过度表达及连锁启动是引发加剧ICM心室舒缩功能障碍的一项重要机制。组蛋白乙酰化修饰中的Ⅰ类HDACs可促进心肌肥厚,如HDAC2可在热休克蛋白70(HSP70)的诱导下被激活,抑制KLFs样因子4(KLF4)、多磷酸肌醇-5-磷酸酶F(Inpp5f)等基因促使心肌肥大。相反,Ⅱ类HDACs已被证明是通过抑制心肌特异性转录因子如MEF2,GATA4和NFAT发挥抑制心肌肥厚的作用〔22〕。HDAC4、5、7和9 能通过抑制转录因子MEF2诱导下游基因的表达,其中HDAC4上结合CaMK的结构域与结合MEF2 的结构域相互交叠, 使得CaMK可以介导MEF2从HDAC上解离, 这表明HDAC-MEF2复合物受Ca2+信号通路中一系列调节因子的控制。但是,现在仍不清楚CaMK/HDAC /MEF2信号通路是否也是心肌细胞分化所必须的〔23〕。美国爱荷华大学研究已证实心肌损伤之后,CaMKⅡ可通过修饰一种特殊的线粒体钙离子通道来调节钙离子进入线粒体,其活性增加过大会增加流进线粒体中的钙离子数量,而这种钙离子超载触发细胞死亡〔24〕,心肌功能下降。
ICM病变复杂多样化,可影响心肌电活动改变心肌细胞膜的通透性,导致胞内钠水潴留。因心肌细胞内无氧糖酵解的增强,细胞内出现酸中毒、细胞外出现高钾,影响心室的除极和复极,使心脏冲动的发放和传导出现异常,引起各种心律失常。HDACs的核易位而发生心肌肥大,导致工作心肌在心肌重构中超极化激活环核苷酸门控4(Hcn4)通道增强子被异位激活,可以使阳离子电流增大,导致致死性心律失常发生,而HDACs抑制剂可激活工作心肌的活动〔25〕。HDAC4从细胞核向细胞质易位可能会引发突触活动;HDAC3受维甲酸和甲状腺沉默调解员激素受体(SMRT)和核受体辅阻遏(N-COR)影响,可促使HDACs发生核异位,激活Hcn4通道增强子,引起 K+、Ca2+、Na+等阳离子电流变化〔26,27〕,从而导致各种心律失常。
2.4组蛋白乙酰化与血管内皮功能失调 ICM血管内皮产生和释放的内源性血管舒张因子一氧化氮(NO)及前列腺素(PGI2)减少,而强有力的缩血管物质内皮素及血管紧张素Ⅱ的分泌增多。内皮祖细胞通过VEGF/PI3K/Akt信号通路分化为成熟的内皮细胞,从而激活HDAC3,HDAC3介导p53蛋白去乙酰化和激活p21。如HDAC3被阻断,通过PI3K-AKT信号通路下调VCAM-1在EC中的表达,影响血管内皮细胞(EC)的形态与存活〔28〕。Ⅱ类HDACs作为调节心血管疾病的关键因素,可抑制MEF2的表达,激活雌激素受体α(ERα)的转录,促进病理性心肌重塑。其中HDAC5、9可上调ER靶基因的血管内皮生长因子α(VEGFα)的表达,促进部分梗死区域的血管生成,从而达到保护缺血心肌的作用〔29〕。白藜芦醇-Sirt1-Foxo1调控通路具有保护缺氧心肌的作用,Sirt1负性转录调节因子Foxo1的转录活性,其下游基因Bim、p27的表达被调控,抑制血管内皮发展。Sirt1内皮特异过表达可激活一氧化氮合成酶(eNOS)的转录,改善血管内皮细胞依赖性主动脉舒张功能,使心肌细胞的细胞周期变长,减少心肌细胞的凋亡〔30〕。
3 小结与展望
ICM是一种常见病、多发病,发病机制涉及心肌供氧需氧平衡失调、心肌细胞能量代谢障碍、缺血对心功能和心肌电活动的影响、血管内皮功能失调等。组蛋白乙酰化在逆转ICM中扮演重要角色,HDACs广泛参与疾病的发生发展,调控心肌供需氧平衡、能量代谢、血管内皮功能等多个方面。ICM的基本病因是冠状动脉动力性和(或)阻力性因素引起的冠状动脉狭窄或闭塞性病变,是一种受多种因素影响的疾病,如高血压、高脂血症、动脉粥样硬化、糖尿病、肥胖症等危险因素。Ⅰ类去乙酰化酶HDAC1、2、3均可上调促炎介质的表达,心脏肥大基因和胚胎期基因活化;HDAC8上调可促心肌肥厚标志性基因心房利钠肽(ANF),参与高血压发病,促进心肌肥厚发展〔31,32〕。Ⅲ类去乙酰化酶的Sirt1参与高密度脂蛋白(HDL) 的合成,改变胆固醇的运输,从而降低冠状动脉粥样硬化的风险;Sirt1 刺激胰腺分泌胰岛素影响血糖代谢,减少了肝脏葡萄糖生成量,有效改善葡萄糖耐量,与胰岛素抵抗相关〔33,34〕,可见组蛋白乙酰化修饰与ICM密切相关,值得深入研究。
除此之外,ICM还与环境因素等相关,且同时受饮食、衰老和遗传等多因素的影响,增加了疾病诊疗的复杂性。研究表明,组蛋白乙酰化、DNA甲基化、组蛋白甲基化和磷酸化等常常共价修饰,故在研究组蛋白乙酰化修饰与ICM时,也应充分结合其他表观遗传修饰调控机制进行研究。通过研究以组蛋白乙酰化为代表的表观遗传调控,能很好地阐释ICM的发病机制,开发更有效的治疗用药。最新研究发现H3K27去甲基化酶UTX在小鼠心脏发育过程中扮演十分重要的角色〔34,35〕,这对表观遗传学在心血管疾病研究领域的应用有着积极的推动作用,如HDACs抑制剂将为临床提供针对性预防和新的治疗靶点〔23〕。中医药能有效预防和改善ICM,而且以组蛋白乙酰化为代表的表观遗传标记物为靶点筛选相应药物有效成分,也将是一个新的研究方向,值得深入研究探索〔36〕。
4 参考文献
1王鸣和,王 俊. 缺血性心肌病的研究近况〔J〕. 国际心血管病杂志,2007;34(1):1-5.
2Chaudhry FA, Iskendrin AE. Assessing myocardial viability in ischemic cardiomyopathy 〔J〕. Echocardiography,2005;22 (1):57.
3Schones DE,Zhao KJ.Genome-wide approaches to studying chromatin modifications 〔J〕. Nature Genetics,2008;3(9):179-91.
4Kouzarides T. Chromatin modifications and their function 〔J〕.Cell,2007;128(4):693-705.
5Schübeler D, Mac Alpine DM, Scalzo D,etal. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote 〔J〕. Genes Dev,2004;8(11):1263-71.
6Minucci S, Pelieci PG. Histone deacetylase inhibitors and the promise of epigenetic(and more)treatments for cancer 〔J〕. Nat Rev Cancer,2006;6(1):38-51.
7Saha R,Pahan K.HATs and HDACs in neurodegenaraion:a table of discencerted acetylation homeostasis〔J〕.Cell Death Different,2006;13(4):539-50.
8Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics 〔J〕. EMBO Rep,2002;3(3):224-9.
9Weichert W, RSske A, Niesporek S,etal. Classl histone deacetylase expression has independent prognostic impact inhuman colorectal cancer:specific role of class I histone deacetylasesin vitro and in vivo 〔J〕. Clin Cancer Res,2008;14(6):1669-77.
10Park JH, Kim SH, Choi MC,etal. ClassⅡhistone deacetylases playpivotalrolesin heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors 〔J〕.Biochem Biophys Res Commun,2008;368(2):318-22.
11Michan S, Sinclair D. Siauins in mammals : insights into their biological function 〔J〕. Biochem J,2007;404(1):1-13.
12De Ruijter AJM,Van Gennip AH,Caron HV,etal. Histone deacetylases (HDACs): characterization of the classical HDAC family 〔J〕. Biochem J,2003;370(Pt3):737-49.
13林琴琴,马 雪,林 蓉.组蛋白去乙酰化酶Sirt1与心血管系统疾病〔J〕.中南医学科学杂志,2011;39(6):601-6.
14王 颖,王生余,侯春梅,等.组蛋白去乙酰化抑制剂SAHA阻断MAPK信号通路并诱导HL-60细胞凋亡〔J〕.中国实验血液学杂志,2007;15(2):267-71.
15Kaneda R, Ueno S, Yamashita Y,etal. Genome-wide screening for target regions of histone deacetylases in cardiomyocytes 〔J〕. Circ Res,2005;97(3):210-8.
16Mano H.Epigenetic abnormalities in cardiac hypertrophy and heart failure 〔J〕. Environ Health Prev Med,2008;13(1):25-9.
17Burch GE, Giles TD, Colcolough HL. Ischemic cardiomyopathy 〔J〕. Am Heart J,1970;79(3):291-2.
18Berry JM, Cao DJ, Rothermel BA,etal. Histone deacetylase inhibition in the treatment of heart disease 〔J〕. Expert Opin Drug Saf,2008;7(1):53-67.
19Shi H, Chen L, Wang H,etal. Synergistic induction of miR-126 by hypoxia and HDAC inhibitors in cardiac myocytes 〔J〕. Biochem Biophys Res Commun,2013;430(2):827-32.
20Pantely GA,Bristow JD.Ischemic cardiomyopathy 〔J〕. Prog Cardiovasc Dis,1984;27(2): 95-114.
21Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure 〔J〕.Expert Opin Ther Targets,2009;13(7):767-84.
22Kemper JK, Choi SE, Kim DH. Sirtuin 1 deacetylase: a key regulator of hepatic lipid metabolism〔J〕. Vitam Horm,2013;91:385-404.
23Olson EN, Backs J, Mc Kinsey TA. Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation 〔J〕. Novartis Found Symp,2006;274:3-12.
24Joiner ML, Koval OM, Li J,etal. CaMKⅡ determines mitochondrial stress responses in heart 〔J〕. Nature,2012;491(7423):269-73.
25Vedantham V, Evangelista M, Huang Y,etal. Spatiotemporal regulation of an Hcn4 enhancer defines a role for Mef2c and HDACs in cardiac electrical patterning 〔J〕. Dev Biol,2013;373(1):149-62.
26Sando R,Gounko N,Pieraut S. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory 〔J〕. Cell,2012;151(4):821-34.
27Morello F, Perino A, Hirsch E.Phosphoinositide 3-kinase signalling in the vascular system. 〔J〕. Cardiovasc Res,2009;82(2):261-71.
28Rooij E,Fielitz J,Lillian B,etal. Myocyte enhancer factor 2 and Class Ⅱ histone deacetylases control a gender-specific pathway of cardio-protection mediated by the estrogen receptor〔J〕. Circ Res,2010;106(1): 155-65.
29王 伟,陈纯娟,傅玉才,等. 白藜芦醇-Sirt1-Foxo1调控通路对缺氧心肌细胞的保护作用〔J〕. 心脏杂志,2009;21(1):23-8.
30Kee HJ, Kook H. Roles and targets of class Ⅰ and Ⅱa histone deacetylase in cardiac hypertrophy 〔J〕. J Biomed Biotechnol, 2011, 2011:98326.
31曹珊珊,李瑞芳,方伟进,等.组蛋白去乙酰化酶8对肾性高血压大鼠心肌肥大的影响〔J〕,中国病理生理杂志,2012;28(2):253-7.
32Li X, Zhang S, Blander G,etal. Sirt1 deacetylates and positively regulates the nuclear receptor LXR〔J〕.Mol Cell,2007;28(1):91-106.
33Chen YR, Lai YL, Lin SD,etal. SIRT1 interacts with metabolic transcriptional factors in the pancreas of insulin-resistant and calorie-restricted rats〔J〕. Mol Biol Rep,2013;40(4):3373-80.
34Lee S, Lee JW, Lee SK,UTX.a Histone H3-Lysine 27 demethylase,acts as a critical switch to activate the cardiac developmental program〔J〕.Developmental Cell,2012;22(1): 25-37.
35Wang C, Lee JE, Cho YW,etal. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity〔J〕.Proc Natl Acad Sci USA,2012;109(38):15324-9.
36卢圣锋,徐 斌,于美玲,等.表观遗传学在中医针灸研究中的应用探讨〔J〕.南京中国医药大学学报,2013;29(2):105-8.
