共缩聚芳砜酰胺结构与性能的研究
2014-08-05李虎敏汪晓峰陈晟晖彭小洁张幼维赵炯心
李虎敏,汪晓峰,徐 兵,陈晟晖,彭小洁,张幼维,赵炯心*
(1.东华大学材料科学与工程学院纤维改性国家重点实验室,上海201620;2.上海特安纶纤维有限公司,上海201419;3.中国科学院上海有机化学研究所,上海200032)
聚芳砜酰胺(PSA)是一种共缩聚物,由4,4'-二氨基二苯砜(4,4'-DDS),3,3'-二氨基二苯砜(3,3'-DDS)以及对苯二甲酰氯(TPC)共缩聚而成,含较高比例3,3'-DDS的PSA溶解性好,而含有较高比例4,4'-DDS的PSA则具有较好的弹性模量及力学强度,但溶解性较差[1]。提高PSA中4,4'-DDS/3,3'-DDS的投料比,将增加 PSA 中的对位结构比例,使PSA具有更加规整的化学结构。由4,4'-DDS与对苯二甲酰氯(TPC)缩聚得到的聚合物称其为全对位PSA,分子结构的对称性高,分子间作用力强,取向性好,由其制备的纤维或膜材料应当具有优良的力学性能;但溶解性较差,PSA聚合物的熔点均高于其分解点,使得这种全对位PSA难以加工。
作者曾研究表征了全对位PSA的结构与性能,采用氯化锂(LiCl)作为助溶剂,与N,N-二甲基乙酰胺(DMAc)组成复合溶剂体系,全对位PSA能够很好地溶解于DMAc/LiCl中,可制得断裂强度达到4.4 cN/dtex的全对位 PSA 纤维[2]。同时引入DMAc/LiCl和DMAc/CaCl2溶剂体系,研究了不同共缩聚比PSA的溶解性能,并通过傅里叶变换红外(FTIR)、核磁共振(NMR)、热失重(TG)、极限氧指数(LOI)、动态热力学分析(DMA)、广角X射线衍射(WAXD)等测试手段分析了不同共缩聚比的PSA聚合物的结构与性能。
1 实验
1.1 原料及试剂
PSA溶液:上海特安纶纤维有限公司和中国科学院上海有机化学研究所提供;DMAc、氯化锂(LiCl)、氯化钙(CaCl2):分析纯,国药集团化学试剂北京有限公司工厂提供。
1.2 试样制备
将PSA溶液涂在玻璃板上刮成膜,然后放入水中,将得到的白色PSA膜经水洗、干燥、研磨制成PSA粉末待用。其中,不同共缩聚比PSA中4,4'-DDS和3,3'-DDS的投料摩尔比分别为 3∶1,4∶1,5∶1 制成的试样,其编号为 1#,2#,3#,全对位PSA为试样4#。
1.3 测试与表征
溶解性能:准确称取一定质量的PSA粉末,加入到盛有溶剂 (DMSO,DMAc,DMAc/LiCl,DMAc/CaCl2)的单口圆底烧瓶中,室温下搅拌4 h进行溶胀,然后用油浴将烧瓶温度加热到40℃进行进一步溶胀或者溶解,4 h后,若是聚合物还没有完全溶解,将温度提高到80℃,使聚合物完全溶解,测试聚合物的溶解性能。
FTIR分析:将少量聚合物剪碎后与适量KBr粉末混合,并研磨压制成薄片,制备成红外试样,用美国 Nicolet公司的NEXUS-670傅里叶变换红外光谱仪进行测试。
NMR分析:用氘代二甲基亚砜溶解PSA,PSA质量分数约为5%,用瑞士 Bruker公司的Avance 400核磁共振仪测试,13C-NMR工作频率为100 MHz,1H-NMR工作频率为400 MHz。
TG分析:取约5 mg的PSA粉末,采用德国Netzsch公司制的209F1 Iris热失重分析仪进行测试。吹扫气为空气和氮气,温度为室温~900℃,升温速率为10℃/min。
LOI:试样制成尺寸为130 mm×6.5 mm×3 mm的薄膜状,采用意大利 Ats Faar公司的FAA临界氧指数仪进行测试,测试方法根据 ASTM D2863—97测试。
DMA分析:采用美国 TA公司的Q800型动态热力学分析仪进行测试,使用薄膜专用夹具,薄膜尺寸为35 mm×6 mm×2.5 mm,温度为30~400℃,升温速率为10℃/min。
WAXD分析:PSA的晶体结构采用日本Rigaku公司D/MAX-2550型X射线衍射仪测试。扫描为5°~60°,电压 40 kV,电流 30 mA,光源CuKα,波长0.154 nm。所得谱图通过分峰软件,可计算出试样的结晶度,由Scherrer公式可计算2θ 在 16.1°的晶粒尺寸[3]。
2 结果与讨论
2.1 溶解性能
从表1可以看出,随着聚合物中对位结构4,4'-DDS比例的增加,其溶解性能逐渐减弱,这与文献[1]的研究所得一致。
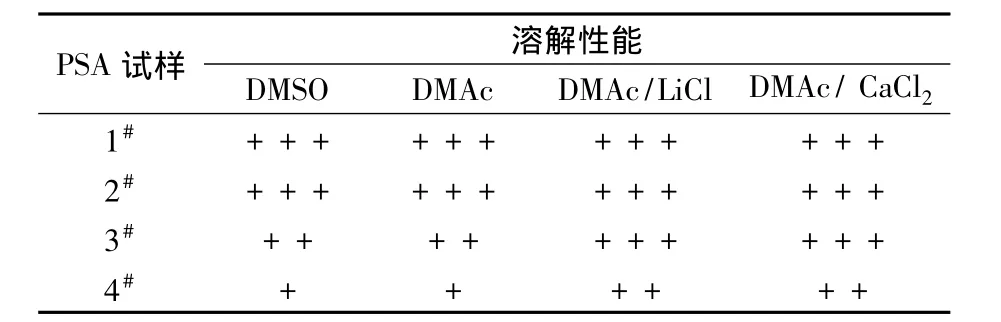
表1 PSA试样的溶解性能Tab.1 Solubility of PSA samples
1#试样的溶解性能非常好,纯DMAc或DMSO在常温下就能将其溶解,3#试样的溶解性能稍稍减弱,在纯溶剂下温度需要达到80℃才能完全溶解,而在DMAc/LiCl和DMAc/CaCl2溶剂体系下还是能够非常容易地溶解。4#试样在纯DMAc和DMSO中不完全可溶,80℃下的溶解度比较小,且小于5%,升高温度也无法提高溶解度,而在DMAc/LiCl和DMAc/CaCl2溶剂体系下,当温度升高到80℃时,全对位PSA均能完全溶解。
2.2 FTIR 分析
从图1可以看出,1 665 cm-1附近为C=O的伸缩振动峰,1 500~1 532 cm-1为N—H弯曲振动和 C—H伸缩振动的组合峰,1 320 cm-1和1 150 cm-1附近两个较强的吸收峰分别对应于砜基(—SO2—)的不对称和对称伸缩振动峰。间位苯基和对位苯基的弯曲振动峰在789 cm-1和837 cm-1处[4],由图1 还可以看出,当 PSA 聚合物中3,3'-DDS比例减少而4,4'-DDS的比例增加时,789 cm-1处的弯曲振动峰的强度降低而837 cm-1处的弯曲振动峰的强度增加。

图1 PSA试样的FTIRFig.1 FTIR spectra of PSA samples
2.3NMR 分析
从图2可见,PSA结构式中各氢(H)原子对应的位置。由于PSA分子链上绝大部分H原子和碳(C)原子都在苯环上,根据取代基团对苯环芳香族H原子和C原子化学位移(δ)的影响,采用文献[5]中公式计算出其δH和δC,可以确定谱图上每一个H原子和C原子所对应的δ。
从图3可以看出,含间位结构的PSA谱图形状类似,但不同峰面积之比有所差别;而4#试样中没有1#试样中所含有的间位结构,所以4#试样的谱图中没有5,6,7,8这4个峰位。

图2 PSA结构式中各H原子位置Fig.2 H atom position in PSA structural formula

图3 PSA试样的1H-NMRFig.3 1H-NMR spectra of PSA samples
通过计算出峰面积,可得到各类H原子的数量之比,得到PSA中对位结构单元与间位结构单元之间的比例。对于全对位的PSA,其各个峰的面积比为S2∶S3∶S4∶2S1约等于1;而对于含间位结构的PSA,对位结构与间位结构之比可以用1号位和5号位的峰面积的关系式(S1-S5/S5)来表示[4],由此可计算出试样 1#,2#,3#的(S1-S5)/S5分别为 2.87,3.85,4.91。从图 4 可见,PSA结构式中的C原子对应的位置。试样1#,2#,3#虽然在聚合反应的过程中,其(4,4'-DDS)∶(3,3'-DDS)摩尔比为3∶1,4∶1,5∶1,但是产物中对位结构和间位结构的比例略小于原投料比,这是计算方法和实验误差造成的,因为最终得到的聚合物浆液中不可能含有未反应的单体存在。

图4 PSA结构式中各C原子位置Fig.4 C atom position in PSA structural formula
从图5可以看出,相比于1H-NMR谱图,13CNMR谱图的峰位较多。4#全对位PSA中没有1#PSA中所含有的间位结构,所以全对位PSA的谱图中没有 8,9,10,11,12,13 这 6 个峰位。

图5 PSA试样的13C-NMRFig.5 13C-NMR spcetra of PSA samples
2.4 LOI
经测试,不同共缩聚比 PSA 试样 1#,2#,3#,4#的LOI均为33%。PSA聚合物的阻燃性能不会随对位结构的增加而变化,间位芳纶的LOI为29%[6],说明PSA的阻燃性能比间位芳纶更好。
2.5DMA 分析
不同共缩聚比PSA在1 Hz下的力学损耗正切(tanδ)与温度的关系由图6所示,图6中全对位PSA的玻璃化转变温度(Tg)在之前的研究中已经报道[2]。
从图6可知,不同共缩聚比PSA的tanδ仅出现一个峰,这个峰即为伴随玻璃化转变的α峰,即聚合物分子链段在此时发生了由冻结到自由的转化,对应的温度即不同共缩聚比PSA的Tg。从图6可知,不同共缩聚比PSA的Tg随着对位结构的增加而小幅度增加,这一结果与Ding Xuan的研究不同[4],Ding Xuan用DSC测得PSA的Tg随着聚合物中对位苯环取代基团比例的增加而有较大幅度增加。由于DMA对Tg的测试精度要比DSC高得多,所以,由DMA测定不同共缩聚比PSA的Tg是正确的。在Tg以下,不同共缩聚比PSA聚合物的tanδ未观察到有明显的出峰,说明PSA聚合物在Tg以下时,几乎没有次级松弛过程,PSA聚合物在玻璃态时,分子链中较小的运动单元[7]不会随着温度的升高而发生从冻结到自由的变化。表明了PSA聚合物高温尺寸稳定性。

图6 PSA试样的tanδ与温度的关系Fig.6 Plots of tanδ versus temperature for PSA samples
2.6 结晶性能
从图7可知,不同共缩聚比PSA聚合物的结晶行为类似,在2θ为22.5°,16.1°左右出峰,而全对位PSA在2θ为16.1°的峰的半高宽明显要小于含间位结构的PSA。

图7 PSA试样粉末的WAXD曲线Fig.7 WAXD spectra of PSA samples
从表2可知,随着PSA中对位苯环取代基团的增加,聚合物的结晶度稍有增长,这表明当PSA逐渐从无规共缩聚物向交替共缩聚物转变时,聚合物的分子结构逐步规整,趋向于紧凑,增进了有序性,使得聚合物的结晶度增加。
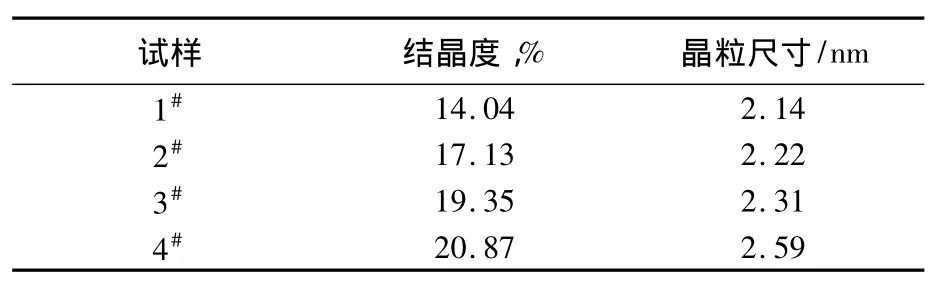
表2 PSA试样的晶体结构参数Tab.2 Crystal structural parameters of PSA samples
2.7 TG 分析
从图8可看出,在氮气氛围中,不同共缩聚比PSA在900℃时燃烧残余量都在40%以上。这说明聚合物中对位结构比例的增加,并不能提高PSA聚合物的热分解温度,在空气和在氮气气氛下,PSA聚合物的热分解性能都相近,这与前人的研究一致[8]。
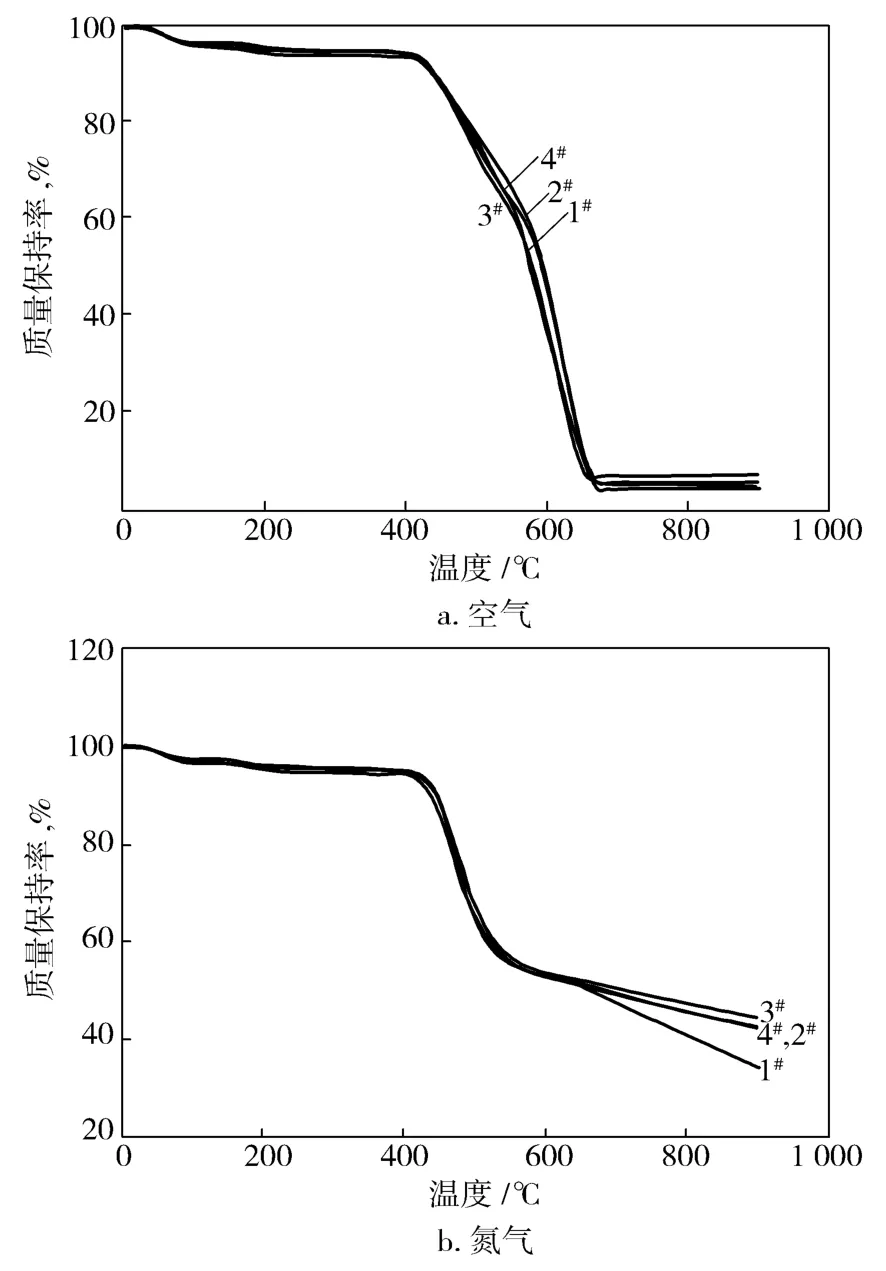
图8 PSA试样在空气和氮气气氛下的TG曲线Fig.8 TG curves of PSA samples in air or nitrogen atmosphere
3 结论
a.随着PSA中对位结构比例的增加,其溶解性能逐渐减弱。1#试样的溶解性能非常好,4#试样即全对位PSA在纯DMAc和DMSO中不完全可溶,而在DMAc/LiCl和DMAc/CaCl2溶剂体系下均能完全溶解。
b.通过 FTIR、1H-NMR和13C-NMR分析对PSA分子结构的表征,发现不同共缩聚比PSA聚合物的分子结构与预期的分子结构基本相同,聚合物中对位结构的比例基本与投料比符合。
c.聚合物中对位结构比例的增加对PSA的热性能和阻燃性能影响不大,但较小地增加了PSA的Tg,不同共缩聚比PSA的LOI都为33%,超过间位芳纶的LOI。
d.不同共缩聚比的PSA聚合物结晶性能都较差,只有在全对位PSA的谱图上能够看到较明显的结晶峰。
[1] 钱勇,黄超伯,丁秋平,等.4,4'-DDS/TPC/3,3'-DDS三元缩合聚合及表征[J].高分子材料科学与工程,2006,22(3):42-45.
[2] Li Humin,Zhu Yin,Xu Bing,et al.Preparation and characterization of all para-position polysulfonamide fiber[J].J Appl Polym Sci,2013,127(1):342 -348.
[3] 常建华,董绮功.波谱原理及解析[M].北京:科学出版社,2010:125-243.
[4] Ding Xuan,Chen Ye,Chen Shenghui,et al.Copolymer structure and properties of aromatic polysulfonamides[J].J Macromol Sci B,2012,51(6):1199-1207.
[5] Pretsch E.波谱数据表:有机化合物的结构解析[M].荣国斌,译.上海:华东理工大学出版社,2002:7-12.
[6] García J M,García F C,Serna F,et al.High-performance aromatic polyamides[J].Prog Polym Sci,2010,35(5):623 -686.
[7] 吴万涛.聚芳砜酰胺纺丝工艺与纤维结构性能的研究[D].上海:东华大学,2010.
[8] 宋江超.芳砜纶纤维的发展和应用[J].国外丝绸,2007,(2):38-40.
