药品数据保护的比较分析与立法选择*
2014-06-21梁志文
梁志文
(南京师范大学法学院,江苏 南京 210023)
一、问题的提出
在知识产权领域,长期存在的争议是如何协调药品可及性与知识产权保护。人们把大量的注意力投入了药品专利保护之上。然而,影响药品可及性的知识产权规则不仅仅来自于专利制度。在国际协议谈判过程中,在人权语境下药品可及性与知识产权保护的所有论争中,很大程度上都忽视了影响药品可及性的另一重要法律制度:数据保护。制药业在TRIPS协议谈判中不仅收获了药品专利保护的重要进展,而且也在第39.3条中获得了额外的重大收益:用于产品上市所递交的数据将在法律上得到保护。所谓数据保护,是指在一定期限内,药品上市主管部门仅允许原研药商使用其递交的、用于证明药品安全性、有效性等信息的数据,药品上市主管部门或者不得允许其他申请者利用生物等效性研究来获得上市申请(这属于较高的保护水平);或者禁止其他人不正当地商业性利用这些数据(这属于较低的保护水平)。
TRIPS协议规定的数据保护不仅成为各成员方所应履行的最低义务,而且也成为后TRIPS时代下制药业在各种国际场合追求更高保护水平的新起点。为落实中国的入世承诺,2002年修订的《药品管理法实施条例》第35条及第72条完成中国的数据保护立法工作,并通过《药品注册管理办法》(2007年修订)第18至20条落实到药品注册的行政保护工作中。①然而,由于对数据保护的具体制度及其影响并未有足够的认识,这些规定与TRIPS协议第39.3条均有一定差距,特别是药品注册的管理工作与其存在一定差距。缺乏有效的数据保护也成为我国连续多年被美国列为《特别301报告》中优先观察国家名录的重要原因。
TRIPS协议第39.3条规定了成员方保护药品数据的义务。但是,成员方应采取何种方式以及在何种力度上保护数据?这在发达国家和发展中国家中产生了争议。因为TRIPS协议本身具有一定的弹性,不同国家采取不同立场,这不仅涉及其制药业研发能力和模仿能力的差异,也与不同国家在处理公共健康问题时所具有的财政能力和技术能力密切相关。在我国,尚未有成果对成员方如何实施TRIPS协议第39.3条予以系统研究。然而,在美国“特别301报告”的压力下,中国必将完善数据保护的法律规则,并强化其法律的执行。如何利用TRIPS协议第39.3条的弹性空间,以构建符合中国国情的数据保护制度,就有必要从WTO成员方实施TRIPS协议第39.3条的不同做法中获得经验。
二、药品数据保护的权利属性
TRIPS协议第39.3条禁止对新型化学成份药品用于上市许可所需的数据进行“不正当的商业利用”;除非出于保护公众利益的需要,或者采取了保证该数据免受不公平利用的措施,不得披露上述数据。该条规定的数据保护,在法律上究竟具有何种性质?不同国家有不同的做法。
(一)禁止占用(商业秘密)模式
发展中国家认为,TRIPS协议第39.3条是以国际贸易中禁止不正当竞争原则来保护数据,它以《巴黎公约》第10条之二为标准,禁止仿制药商以不正当手段获取品牌药商用于申请上市的数据加以商业性利用,或为获取不当的竞争优势而窃取数据。此为数据保护的禁止占用模式,它允许竞争者通过反向工程等方式获取上市所须的数据。本质上,它是有利于仿制药尽快入市的一项数据保护制度。[1]P167
数据保护的禁止占用模式具有四项基本特征:[2]P272一是从保护对象看它仅保护未公开的数据,即须符合秘密性特征;二是国家药品主管当局对递交其审批的未公开数据负有保密义务;三是法律禁止仿制药商以不当方式获取或利用这些数据;四是国家药品主管当局可自由依据品牌药商递交的所有药品数据来审批仿制药的上市申请,无须等待法定期限届满,这不属于“商业性利用”数据行为。
埃及《知识产权法典》(2002)在第一编第三部“未公开信息”(即商业秘密)中提供药品数据的保护。其第56条规定:“依据本法相关条款规定,本法提供的保护延及向有关当局提交的、用于获取产品上市审批的未公开信息,其条件是,包含有新型化学成分的药用化合物或农用产品,且其获得需要付出重要努力。受理这些信息的有关当局有义务予以保护,不得披露,并禁止非法的商业使用行为;其保护期限自信息递交之日起直至其不再符合秘密的条件,或最长不超过5年,以两者更短为准。有关当局为保护公众利益而公开信息,不属于侵犯信息所有人的权利。”此外,第56条规定,该法“相关条款”也适用于数据保护。故而,在埃及法上,数据保护不包括有关当局利用品牌药商递交的未公开数据来审批具有生物等效性的仿制药上市申请。
阿根廷也以商业秘密的方式保护药品数据,且没有规定数据保护的法定期限。《秘密信息和产品法案(第24766号)》第4条规定:“在阿根廷或其他国家境内未被注册的使用了新型化学成分的产品,其安全性和有效性信息在递交公共卫生主管当局之后,如果其符合第1条(商业秘密)规定的条件,且其获取须付出重要的技术或经济努力,不得予以披露,以及禁止本法所指的不诚信的商业利用行为。”因此在阿根廷,“仿制药商可以递交生物等效性数据,即,与药品监管部门已批准上市的原研药具有实质相似,就可以获得仿制药的上市许可。仿制药商既不需要对品牌药商予以补偿,也无须取得其同意。尽管仿制药商可能会面临专利的问题,但其在获得上市许可之前仍无须任何等待。”[3]P153
概括说,禁止占用模式下的数据保护以不正当竞争(商业秘密)为中心,具有示范性的立法通常表述为:“政府当局禁止非法占用为获得包含有新型化学成分的药品或农用品上市而递交给当局的未公开数据或其他数据。除非为保护公共利益所必须,政府当局不得予以披露。”[3]P154采用这种立法模式的还有南非、印度、土耳其等,它们均允许仿制药商依据品牌药商递交的数据、以生物等效性而申请上市。[4]P131
(二)数据专有权模式
专有权模式为美国所推崇。专有权模式强调,对数据的不当利用既包括仿制药商采取不当手段获取这些数据并将其用于仿制药上市的审批之中,即直接利用数据的行为;也包括有关当局利用这些数据,通过评估仿制药与原研药的生物等效性,从而批准仿制药的上市,即间接利用数据的行为。[5]P527-529从数据保护的权利内容来看,专有权模式可分为两类:数据专有权与市场专有权。市场专有权模式后文讨论。
数据专有权是指在一定保护期限内,仿制药商不得依赖品牌药商所递交的数据,即不得通过生物等效性研究来获得仿制药的上市许可;主管部门也不得依赖这些数据来审批仿制药的上市。但是,如果仿制药商独立投资研究获得药品上市许可所需的药品安全性、有效性和质量可靠性的数据,既使处于法定数据保护期内,仿制药商也可提出上市申请,主管部门也可根据仿制药商独立获得的数据批准上市申请。②
数据专有权模式也包括两种不同的立法例。
(1)以公开为基础的立法例。早期的美国《联邦食品、药品与化妆品法》(FDCA)将品牌药商递交的数据视为其商业秘密而给予保护,并且没有保护期限制;FDA负有保密义务,直至其不具有秘密性为止。1984年Hatch-Waxman法案建立了数据的专有权模式,而不是商业秘密的保护方式。该法案的目标之一是鼓励仿制药商的上市,其重要变革是数据的可公开性。即,FDA对品牌药商递交的数据不负有保密义务,未公开的安全性、有效性数据或其他与药品有关的信息均可由第三方依据《信息公开法》的申请而向其提供,除非属于特殊情况。③依据FDCA第505条的规定,④“当新药申请(NDA)不被批准或被放弃,或者依据原研药的研究数据来审批仿制药时,或者于仿制药将被批准上市的最早日期,品牌药商递交的研究数据就可以被公开。质言之,当研究数据不再被构成竞争性障碍时,其必须被公开。从立法语言层面来看,人们可以推断,国会已经解决了(数据披露的)问题:只要没有相反规定,所有的研究数据就应能够为公众所获得。”⑤
(2)以保密为基础的立法例。在1987年以前,欧盟以商业秘密的方式来保护药品数据,且各国的保护标准和力度各不相同。数据保护的现行有效法律为《第2001/83/EC号指令》(为《第2004/83/EC号指令》修订),它赋予品牌药商自授权日起10年(8+2+1)专有权保护。在第8年保护期届满之后,只要仿制药商能够生产与原研药“实质相似”的药品,药品主管部门可以依赖品牌药商所递交的数据来快速审批仿制药的上市。但是,依据该指令,主管部门在任何时间都不得向公众公开测试数据。[2]P271新西兰也采保密方式保护药品数据,并规定了主管部门的保密义务,只有在特殊情况下才可以公开这些数据。新西兰1981年《药事法》第23B条规定,主管部门对创新药申请时所递交的保密信息,在5年保护期内,应采取合理的措施予以保密,且不得利用秘密信息来审批其他上市申请。同时,第23C条规定了三种情况下可以向他人披露或使用保密信息。⑥
从立法模式来看,我国法采取以保密为基础的数据专有权模式来实施TRIPS协议第39.3条。依据《条例》第35条之规定,我国法禁止对新型化学成份药品数据进行“不正当的商业利用”,而“不正当的商业利用”是指“不依赖”、“不披露”,即,未经原研药商之同意,不批准利用数据而“申请生产、销售新型化学成分药品的许可”;除非符合法定条件,不得披露上述数据。但是,仿制药商提交自行取得的数据,并不受法律的限制。
数据专有权保护还有一种特殊的制度:为儿童用药品的研发提供额外的激励。美国在1997年通过《药品和食品管理现代化法》,为激励制药企业投资开发儿童用药,对获取儿童用药上市许可的制药企业提供额外的6个月数据保护。这6个月的数据保护并不伴随对儿童用药的上市许可而授予,也不限于该类用途。其获取的唯一条件是:属于FDA所规定的儿童用药研究,并在FDA所规定的时间框架内完成研究并递交相关数据。而且,授予6个月的数据保护期不考虑儿童用药研究的结果是正面还是负面的效果。其保护期的起算并不是从获得FDA上市许可之日起立即开始,它是对数据递交者所拥有的其他专有权保护期限的一种延长。在这延长的6个月内,FDA不得批准竞争性的仿制药上市。⑦
欧盟也在2006年通过了儿童用药的指令(1901/2006/EC),[6]其关于数据保护的条款也与美国法极为类似。依据该指令第36条,专利权人或辅助保护证书的权利人可以获得6个月的保护期延长。其条件是:在递交上市许可申请书的产品特征摘要中写入儿童临床试验数据,且其明确为特定儿童人群所进行的临床试验。儿童用药的临床测试证据作为药品上市申请的一部分而提出,而其保护期延长被视为是对儿童用药临床研究所付出的额外成本之补偿。药品制造商可以自行对已有药品的儿童用处方进行临床测试;也可应请求而测试儿童用药,但儿童用药委员会有权决定何种药品开发儿童用药。
我国非常缺乏儿童用药品。我国目前有3500多种化学药品制剂,可供儿童专用的不足60种;国内市场90%的儿童使用的是成人药物的减量版。这导致“儿科医生开药时心都是慌的,”因为“说明书上大多写‘儿童酌情减量或减半’,‘酌情’意味着没有经过儿童临床研究,给儿童使用很不安全,药品过敏的几率很大。”[7]但是,我国并没有儿童用药数据保护的制度。因此,从制度上激励儿童用药临床研究是非常有必要的。因为这是美国立法的经验总结。在美国,“从为儿童标签药品开展试验的角度来看,儿童用药专有权项目已经取得了成功。自1997年项目运行以来,超过115项产品标签有了(儿童用药的)变化。其中,约有三分之一的药品标签变化表明,儿童用药在剂量、安全性和有效性方面与成人患者用药有重要差异。这些新信息有益于儿童的长期健康。”[8]P487
(三)市场专有权模式
市场专有权模式是指在专有权保护期限内,药品主管部门不得批准品牌药商之外的同一药品或类似药品的上市许可,无论用以申请上市的数据是仿制药商自己独立投资开发还是通过其他途径获得。许多国家为激励罕见病药品的开发而提供市场专有权模式的保护。我国尚无明确的法律界定罕见病,但根据世界卫生组织千分之零点六五到千分之一的定义,国内罕见病患者人数高达几千万,这些病患常常因缺乏必要的药品或因需要特别定制药品而支付昂贵的药费。如果一项药品同时符合新药申请的条件,也符合罕见病药品申请的条件,则市场专有权模式提供的是极其类似于专利保护的一种专有权。很明显,它比数据专有权的保护力度更大。[9]P657
以美国1983年孤儿药品法案为例,为解决罕见病治疗药品缺乏足够市场利润的问题,该法案建立了四项激励机制:⑧(1)FDA对临床试验提供辅助性服务。FDA和开发者共同合作确立必要的临床试验,并辅以快速审批机制以尽快地将罕见病药品推向市场。(2)临床试验成本的税收优惠。罕见病药品开发者可将其成本的50%作为税收减免,从而直接降低了开发成本。(3)联邦奖励。对于已有药品的未批准用途或未批准的新药进行临床试验,给予每年10万至20万美元的奖励,并每三年重新评估并予以再奖励;现每年所支付的奖励金额在2500万美元左右。(4)7年的市场专有权。这是对罕见病药品开发最为重要的激励机制,因为它使得开发者获得类似于专利的市场垄断。当然,罕见病药品的市场专有权在权利范围、行使条件等方面,也有不同于传统药品或方法专利权的地方。[10]P131
自2006年以来,我国有人大代表、政协委员呼吁国家应尽快为罕见病立法,使防治工作有法可依、获得保障。[11]尽管我国法律对罕见病药品开发提供了快速审批机制,但并未有任何法律为其开发提供特别的激励机制。应该建立多重的法律机制,也有必要建立罕见病药品数据的市场专有权模式。
三、药品数据受保护的条件
数据受保护的条件与药品是否受专利保护无关。药品数据受保护的条件包括:(1)用于上市许可所需的数据;(2)未披露的数据;(3)使用新型化学成分的药品(NCEs数据专有权),或者具有新用途的药品(辅助性数据专有权),或者首次开发出适用于儿童的药品(儿童用药保护);(4)需要付出相当程度的努力才能获取。在这些条件中,新型化学成分的界定最为重要,且在不同国家具有不同做法,因此是完善数据保护的关键制度之一。
(一)“新型”化学成分的界定
尽管不同于专利法上的新颖性条件,新型化学成分也存在三种不同立法的选择:绝对新颖性(即全球范围内未被披露)、地区新颖性(如欧盟境内首次申请的标准)以及相对或本地新颖性。绝对新颖性是指药品中包含的新型化学成分(新用途、儿童用药等)如果一旦向任何国家的主管部门递交上市申请或其申请已被批准,该化学成分就属于已知或旧的化学成分。例如,阿根廷就采取这一立法模式。⑨
美国法采取相对新颖性的标准,即,未曾被FDA批准上市的药品所包含的化学成分就是新型化学成分。⑩美国等发达国家大都采取相对新颖性的标准,并通过自由贸易协定等向发展中国家推广。我国现行法并没有界定“新型化学成分”,但对“新药”的界定采纳相对新颖性的标准,即“未曾在中国境内上市销售的药品。”在这一背景下,我国法对“新型化学成分”也有可能采纳这一标准。但是,由于含有新型化学成分的药品很少由我国药企研发,相对新颖性标准的采纳就有可能使得外国药企能够最大程度的延长其保护期。例如,在NCEs受专利保护之情形下,原研药商就有可能拖延该药品在我国的上市申请,最大可能地获取药品的市场垄断。为了解决发展中国家的药品可及性问题,被称之为“机会窗口”或“等待期”的制度就为许多发展中国家所采纳。[12]P332-333
机会窗口制度类似于专利申请中的国际优先权制度,是指在国外首次获得上市许可的药品所包含的化学成分仅在法定期限内向本国提出上市申请,才符合NCEs的要求。采取这种立法的国家有智利、以色列和约旦。我国台湾地区《药事法》(2005年)第40-2条第3款规定:“新成分新药在外国取得上市许可后3年内,必须向中央卫生主管机关申请查验登记,始得准用第二项(即5年数据保护期)之规定。”等待期制度是对数据专有权保护期限的限制制度,是将数据专有权的保护期期限之起算点以国外首次获得上市许可之后某一法定期限届满之日,它并不影响NCEs地位的认定。马来西亚、土耳其以及秘鲁就是采取这一种立法模式。
就我国而言,机会窗口制度的立法模式更值得借鉴。其理由有二。第一,绝对新颖性的立法过于刚性,原研药商可能有正当理由(如本地化临床试验等)延误上市申请,如果因此而丧失权利保护,则不甚公平;同时,也易于与美国发生纠纷。例如,台湾地区修订前的药事法规定就引发了美国制药业协会的不满,成为其要求将台湾地区纳入特别301观察国家(地区)的重要理由。第二,将数据保护的期限计算从国外批准上市之日起计算,并不影响原研药商的数据专有权保护地位,同时又有力地预防了原研药商利用数据保护来实现其最长期限的市场垄断,从而影响药品可及性的不当策略。此外,规定合理的等待期限(如12个月)是非常必要的,这是原研药商为满足本地药品上市条件而开展必要试验所必须;如果本地主管当局对上市许可产生了过分迟延,对原研药商给予一定补偿也是合理的。
(二)“化学成分”与“新用途”的界定
美国法区分新型化学成分药品和新用途药品而给予不同的数据保护。对于缺乏创新能力的发展中国家,有学者建议严格限定保护对象,以保障仿制药的市场进入,从而实现药品可及性的目标。这一观点反对新用途药品的数据保护,因为它并非实施TRIPS协议第39.3条所必需;同时主张严格限定NCEs的范围。[13]P271-272我国医药产业具有一定创新能力,特别是模仿创新战略成为我国医药产业发展的重要举措。因此,建立新用途药品的数据保护是合理的。但是,也必须认识到数据保护不仅是激励创新的重要制度,而且也是严重影响公共健康的制度。
美国法对NCEs给予5年的数据保护,对于其他新药给予3年的辅助性数据保护。Hatch-Waxman法案并未使用新型化学成分的术语,而是活性物质的术语。FDA制定的相关条例将“活性物质”界定为 “未被批准药品所包含的活性物质”解释为“包含有新型化学成分的药品”,并将“新型化学成分”界定为“由FDA批准的任何新药申请中未曾包含的活性基。”“活性基”是指“药品成分中有生理或药理作用的分子或离子,而不包括使药品成酯、成盐(包括含有氢键或配位键的盐),或者分子的其他非共价键衍生物(例如络合物、螯合物或包合物)”。FDA对新型化学成分的解释得到了法院的支持。
辅助性数据专有权在欧盟法上仅包括新适应症,而美国法还包括新适应症、新盐、新酯以及新复方;欧盟法对新适应症的开发采延长原专有权的做法,而美国法仅对新适应症予以保护,不延及原专有权。加拿大以“创新药”和“新药”来区分不同的数据保护。“创新药”指“未曾为已批准药品中所包含的药用成分,也不是已批准药品中药用成分的变化,如盐、酯、对映体、溶剂化物或多形体。”“新药”则包括在加拿大未曾批准的新临床适应症、给药形式、处方形式或其他变化。
我国应严格限定NCEs的范围,但适度放宽新用途药品范围。可借鉴发展中国家界定NCEs范围的做法,例如,智利和秘鲁就严格限定于具有药用效果的活性基,而不考虑其不同形式、表达或处理等,并明确规定不属于NCEs的情形。智利法第90条第2款规定:“下列情形均不得视为NCEs:(1)与已予卫生注册或已获许可的同一化学成分具有明显不同的用途和治疗适应症;(2)与已予卫生注册或已获许可的同一化学成分具有不同的给药途径或剂型;(3)对已获授权或注册的化学成分在药用形式、复方或配方的变化;(4)在已予卫生注册或已获许可的化学成分的基础上不同的盐、络合物、结晶形式或化学结构。”但是,与发展中国家仅将新适应症视为辅助性专有权保护的对象不同,我们认为不属于NCEs、但具有明显临床益处或在安全性和有效性方面具有明显进步的药品纳入辅助性保护的对象。即,新适应症、改变给药途径或释药方式的新剂型,如缓释制剂(脂质体、微球、微乳等注射液)等药品。
药品保护还与递交上市许可所须数据相关。美国有些签订的FTA甚至对未曾提供审批所需临床试验数据的药品也要求予以保护,但这不属于TRIPS协议第39.3条的义务。我国能够独立承担药品上市审批的任务,故不应将其作为数据保护的条件。
四、药品数据保护的期限
数据专有权保护的力度大小主要体现在其保护期的长短上。各国法上保护期限的长短具有一定程度的任意性,并无绝对说服力的理论和事实依据。
对于以仿制药的生产和销售为主、处于模仿创新阶段的我国来说,在符合国际条约义务的前提下,应尽量降低数据专有权的保护期限,以促进仿制药的上市,从而提升药品供给的市场竞争、保障药品的获取。《中国加入WTO工作组报告》对NCEs药品承诺了6年的保护期,因此,这是我国法的最低保护水平。但我国对数据的保护期限立法仍然存在自由度。例如,对于保护期的起算方法。为了防止原研药商利用数据保护来获取最长的市场垄断,其起算方法可以规定为:数据保护的期限自药品被批准上市之日起计算;但药品已在他国获准上市,如果自最早获准上市之日起超过一年后才在我国提出上市申请的,则自首次批准上市之日起计算。
表1 各国法上药品数据保护的专有期限
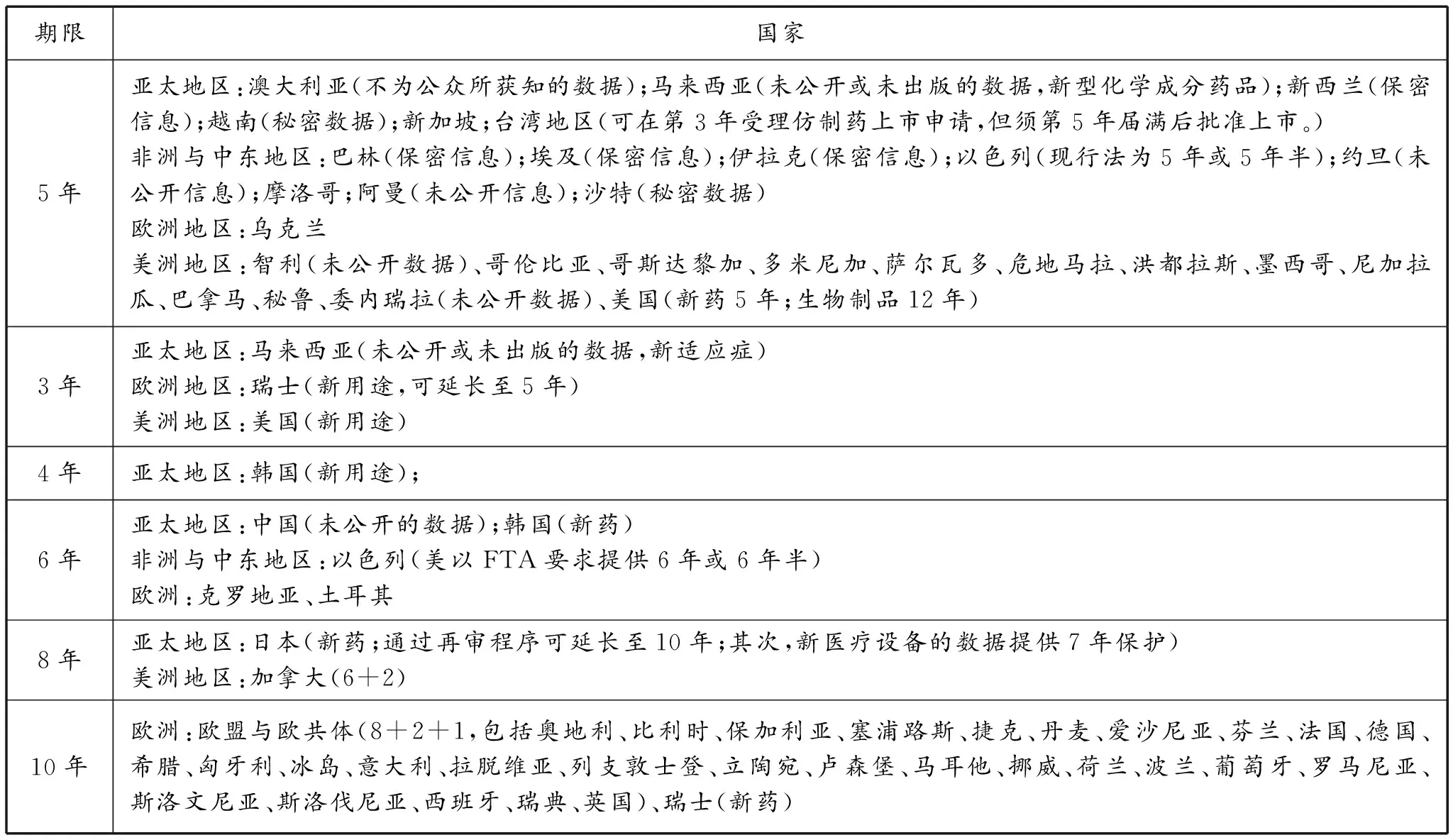
表1 各国法上药品数据保护的专有期限
期限国家5年亚太地区:澳大利亚(不为公众所获知的数据);马来西亚(未公开或未出版的数据,新型化学成分药品);新西兰(保密信息);越南(秘密数据);新加坡;台湾地区(可在第3年受理仿制药上市申请,但须第5年届满后批准上市。)非洲与中东地区:巴林(保密信息);埃及(保密信息);伊拉克(保密信息);以色列(现行法为5年或5年半);约旦(未公开信息);摩洛哥;阿曼(未公开信息);沙特(秘密数据)欧洲地区:乌克兰美洲地区:智利(未公开数据)、哥伦比亚、哥斯达黎加、多米尼加、萨尔瓦多、危地马拉、洪都拉斯、墨西哥、尼加拉瓜、巴拿马、秘鲁、委内瑞拉(未公开数据)、美国(新药5年;生物制品12年)3年亚太地区:马来西亚(未公开或未出版的数据,新适应症)欧洲地区:瑞士(新用途,可延长至5年)美洲地区:美国(新用途)4年亚太地区:韩国(新用途);6年亚太地区:中国(未公开的数据);韩国(新药)非洲与中东地区:以色列(美以FTA要求提供6年或6年半)欧洲:克罗地亚、土耳其8年亚太地区:日本(新药;通过再审程序可延长至10年;其次,新医疗设备的数据提供7年保护)美洲地区:加拿大(6+2)10年欧洲:欧盟与欧共体(8+2+1,包括奥地利、比利时、保加利亚、塞浦路斯、捷克、丹麦、爱沙尼亚、芬兰、法国、德国、希腊、匈牙利、冰岛、意大利、拉脱维亚、列支敦士登、立陶宛、卢森堡、马耳他、挪威、荷兰、波兰、葡萄牙、罗马尼亚、斯洛文尼亚、斯洛伐尼亚、西班牙、瑞典、英国)、瑞士(新药)
数据专有权保护首次获准新药上市的原研药商,且应允许其转让。该权利的重要内容是禁止其他申请者未经其同意而依赖生物等效性研究提出仿制药的上市申请,主管部门既不能受理也不得批准该申请。但是,数据专有权并不禁止其他申请者独立进行临床试验获取上市许可所须的数据,并获得批准。同时,为了促进仿制药的尽快入市,我国法应借鉴欧盟、加拿大以及我国台湾地区的立法,规定在一定期限届满后可以提出生物等效性的上市申请。本文建议:对NCEs新药,自获准上市之日起4年后,其他申请者可提出生物等效性的上市申请,符合条件的,将于NCEs新药获准上市6年届满之次日起批准;对于其他新药,则自获准上市之日起2年后,其他申请者可提出生物等效性的上市申请,符合条件的,将于该新药获准上市4年届满之次日起批准。
五、药品数据保护的限制
药品数据保护制度激励药品创新,但同时也应促进药品可及性以保障公共健康。发展中国家反对数据专有权的保护模式,重要理由是担心该制度将影响公共健康。各国在药品数据保护的立法上规定了不同的限制制度,它们包括如下基本规则:
第一,原研药商的实施义务。这些立法既包括发达国家,也包括发展中国家。例如,加拿大法规定“如果创新药并未在加拿大销售”,则创新药保护的规定就不予适用。沙特阿拉伯法规定,“在王国首次注册的产品被批准上市之后,如果在注册主管当局决定的合理期限内未能推向市场,”该当局在保护期内可允许第三人使用这些未经披露的测试数据。智利法第91条第3款规定,“自注册或批准上市之日起12个月内未能将药品或农用化学产品商业化的,”数据保护就不再适用。哥伦比亚法第4条第4款也有类似规定。这些做法值得我国借鉴。
第二,基于公共健康的限制。TRIPS协议承认药品创新保护与公共健康两者之间需要通过权利限制来协调,这也同样在美国签订的部分FTA中得到承认。因此,应该将基于公共利益而限制数据保护的情形法定化。这些立法也同样包括发达国家和发展中国家。加拿大法规定,依据第C.07.003条(《加拿大药品可及性机制》)而递交的新药申请,构成对创新药保护的限制。具体来说,基于公共健康的限制包括:(1)强制实施许可而对数据专有权的限制。如智利法第91条第2款规定,“符合本法而颁发的强制许可所包括的药品或农用化学产品,”其数据保护就不再适用。马来西亚法第5条第1款也有类似规定。哥斯达黎加法第10条规定了健康主管当局可颁布非排他性的强制许可,也包括基于严重影响公共健康情形下的强制许可,但需要支付合理补偿。(2)基于公共健康而由政府采取的合理措施。马来西亚法第5条第2款、智利法第91条第1款、哥伦比亚法第4条第3款也有类似规定。(3)平行进口对数据保护的限制。美国FTA中大都限制了专利药品的平行进口,但并未涉及数据保护的平行进口问题。我国法允许专利产品的平行进口,也应同样允许数据保护的平行进口。特别是有些药品不受专利保护,允许平行进口将提高药品的市场竞争,从而保障药品可及性。
第三,政府使用或公共利益使用的限制。药品的安全性或有效性事关公共利益,对这些数据的使用属于评估药品的必要措施。不仅主管部门需要使用这些数据,而且其他政府部门、非政府组织和国际组织出于监督药品评估的原因也可能需要使用这些数据。这些对数据的利用,本身就不属于对数据的“商业性利用”。例如,新西兰药事法第23C(1)(b)以及(c)条规定了保密信息向政府有关部门、非政府组织和国际组织公开不属于数据保护的范围。
第四,数据透明度和公众参与程序。审批信息的透明度不仅保护原研药商的创新活动,也是公众获取药品信息的重要途径。美国已建立了官方的数据公开数据库,[14]P137我国现行法并没有类似规定。鉴于数据公开须依托于该数据库,建议参考专利授权数据库的方式,在剔除与药品安全性、有效性和质量可靠性无关的信息之后,向公众开放查询。此外,为监督药品评估工作,公众参与程序的构建是非常必要的。一方面,公众对于主管部门的数据专有权决定不服的,应该可以复议程序来解决争议;对复议决定不服的,可依行政诉讼的方式来裁决。另一方面,原研药商也需要该程序来保障自己权益。
第五,仿制药的快速审批程序。激励仿制药的尽快上市具有重要的公共健康意义。在美国,四分之三的药品为仿制药,它每年为医疗保障体系节约了高达193亿美元的医药费用。这是Hatch-Waxman法案的重要成效,其原因是该法案为仿制药上市提供了快速审批程序。依据该程序,仿制药商无需提供药品的安全性、有效性信息,仅须通过生物等效性研究即可,这节省了仿制药商的大量成本。[15]当生物制品日益成为重要的医药产品时,美国在对生物制品给予更强的数据保护时,也同样为仿制药上市提供了快速审批程序。依据该程序,仿制药商也仅须通过生物相似性研究即可获得上市批准。[16]这同样值得我国借鉴。
第六,Bolar例外。尽管我国专利法在2008年修订时规定了Bolar例外,但《药品注册管理办法》规定的Bolar例外明显不符合我国创新实践的需要。因此,需要在《药品管理法》层次规定该规则。借鉴我国台湾地区药事法第40-2条第3款,我国法应规定:“新药专利权不及于药商在药品申请上市许可过程中进行的研究、教学或试验行为。”
第七,激励仿制药商质疑专利权效力的机制。药品专利无效将为仿制药上市扫清重要的法律障碍。美国法为激励仿制药商挑战药品专利权的效力,法律规定成功挑战专利权效力的仿制药商将获得6个月的专有权期限。尽管该项制度在美国存在争议,但促进仿制药上市的基本目标却已基本实现。我国也有必要予以借鉴。
结论
《中国加入WTO工作组报告》第284段就如何实施TRIPS协议第39.3条这一问题做出了相应的承诺,即为药品提供6年的数据专有权保护。这一立法模式是巴西、印度等发展中国家所反对的,原因是发展中国家普遍缺乏原研药的研发能力,数据保护严重影响其仿制药的上市。我国也是以仿制药为主但具有一定模仿创新能力的发展中国家。《报告》第284段选择了以高水平的数据专有权模式来落实第39.3条,承诺的做出是匆忙的。
幸运的是,与TRIPS协议一样,《报告》第284段仍然有大量的弹性空间可供操作,这些弹性空间可以通过立法来调节数据保护的强度,以适应中国的药品创新实践。我国法对于数据保护存在体系混乱、缺乏合理的执行力度之缺陷;制度本身也过于粗疏,立法层次过低,缺乏对公共健康保障的必要机制,特别是保密为基础的数据专有权模式有可能产生严重的不利后果。因此,有必要重构我国法上的数据保护体系,完善数据保护的基本制度,以实现药品创新激励和公共健康保障的法律目标。同时,由于数据专有权的保护事关药品创新激励与公共健康保障之关系,应该将其法律规定上升至《药品管理法》的层面,而不应由其《实施条例》来担当。当然,还须重视数据专有权保护对公共健康的影响,它需要数据保护的限制规则来予以保障。这是构建具体的制度时应予以考虑的重要事项。
注释:
① 我国对药品数据保护的入世承诺及其保护模式,参见梁志文:《药品数据的公开与专有权保护》,《法学》2013年第9期。
② 美国法上,此申请方式系依据21 U.S.505(b)(1)条所提出的所谓“完整或独立申请”。另外,“值得注意的是,第二个进入市场的药商独立获取相关数据并没有为第三个及其以后进入市场的药商打开审批的方便之门。因为主管当局不得依据第二个药商所递交的数据来审批第三个进入者的上市许可,即使第二个进入者的数据并不受法律保护。其原因是,如果允许第三个进入者依赖第二个进入者所递交的数据来审批仿制药的上市,则品牌药商数据专有权保护的有效期限将大为缩短。”See Valerie Junod, Drug Marketing Exclusivity under Unites States and European Union Law, 59 FOOD DRUG L.J. 479, 492 (2004).
③ See Shreya Matilal, Do Developing Countries Need a Pharmaceutical Data-Exclusivity Regime? 32(6) E.I.P.R. 268, 270 (2010).但是,美国FDCA第331(j)条原则上禁止公开商业秘密;FDA的相关条例也规定在公开某些信息时,应考虑“商业秘密人的产权”。See Elizabeth A. Rowe, Striking a Balance: When Should Trade Secret Law Shield Disclosures to the Government? 96 IOWA L.REV. 791, 808-9 (2011).关于生物制品上市许可中的商业秘密保护,see Richard A. Epstein, The Constitutional Protection of Trade Secrets and Patents under the Biologics Price Competition and Innovation Act of 2009, 66 FOOD DRUG L.J. 285 (2011).
④ 该条原文翻译如下:“除非在特别环境下,依据(b)节申请所递交的安全性、有效性数据和信息须依请求而能够为公众所获得:(A)不再有审批申请案的任何工作进行或即将进行;(B)如果秘书长依据决定申请案不被批准,且所有法律上诉救济均已穷尽;(C)如果申请案的批准文件……已被撤销,且所有法律上诉救济均已穷尽;(D)如果秘书长已经认定涉案药品不属于新药;或(E)对于本节(j)款所指的药品,自首次申请批准的有效日期起;或者对于本节(j)款所指的药品之上市申请是有效的,如果这样的申请已经递交,则自其申请被批准之日起。”
⑤ See Mustafa Unlu, It Is Time: Why the FDA Should Start Disclosing Drug Trial Data, 16 MICH. TELECOMM. TECH. L. REV. 511, 524 (2010).该文指出,产业界将Hatch-Waxman法案视为是未经处理的创设数据公开体制(disclosure regime)的企图。在该法案通过的最后一刻,可能是因为立法者对于公开政策中商业秘密的性质和地位具有根本性的分歧,在立法中插入了数据公开的例外规定。
⑥ 这两款系1994年《药事法修正案》(1994第128号法案)第2节所引入,并于1995年1月1日生效。
⑦ See 21 U.S.C.§355a. 该条原定于2012年失效,但美国国会已经延伸了该法的效力, 并通过了两部立法:《2002最佳儿童用药品法》和《2003儿童用药研究平权法》。前者延长了儿童用药数据专有权条款,并扩展到非专利药的开发;后者授权FDA在特定情况下强制要求制药企业提供儿童用药,从而改变了过去自愿参与儿童用药的做法。更详细的论述,see Govin Permanand et.al., The EU's New Paediatric Medicines Legislation: Serving Children's Needs?92(9) ARCH DIS CHILD. 808, 809 (2007).
⑧ 美国法上更详细的介绍,see David Loughnot, Potential Interactions of the Orphan Drug Act and Pharmacogenomics: A Flood of Orphan Drugs and Abuses? 31 AM.J.L.& MED. 365, 370-372 (2005).
⑨ 《阿根廷秘密信息和产品法》(No.24,766号法令)第4条规定:“申请注册或上市许可的产品包含有在阿根廷或其他任何国家未曾注册的新型化学成分,递交给当地公共健康当局的有关产品安全性和有效性信息,只要符合本法第1条的规定,且需要付出技术和经济的努力,它将受本法规定的禁止不诚信商业利用的保护,且不得披露。”
⑩ See 21 U.S.C 355(j)(5)(F)(ii).
参考文献:
[1] UNCTAD ed., Using Intellectual Property Rights to Stimulate Pharmaceutical Production in Developing Countries: A Reference Guide[M]. New York: United Nations, 2011.
[2] Shreya Matilal, Do Developing Countries Need a Pharmaceutical Data-Exclusivity Regime? [J].32(6) E.I.P.R. 268, 272 (2010).
[3] Robert Wissman, Data Protection: Options for Implementation[A]. in Negotiating Health: Intellectual Property and Access to Medicines [C](Pedro Roffe et.al. eds. ), London: Earthscan, 2006.
[4] Ingo Meitinger, Implementation of Test Data Protection According to Article 39.3 TRIPS: The Search for a Fair Interpretation of the Term “Unfair Commercial Use” [J]. 8 (2) J. WORLD INTELL. PROP. 123, 131 (2005).
[5] Razvan Dinca, The “Bermuda Triangle” of Pharmaceutical Law: Is Data Protection a Lost Ship? [J] 8 (4) J. WORLD INTELL. PROP. 517, 527-529 (2005).
[6] Montserrat Lopez-Bellosta &Ana Beneto Santa Cruz, Fostering Pediatric Research and the Right to Extend Supplementary Protection Certificates[J]. 5 J. INTELL. PROP. L & PRACTICE 45 (2010).
[7] 薛冰妮、彭文斌.儿童吃成人药,医生都心慌[N].南方都市报2012年-12-3(A08).
[8] Jennifer S.Li.et.al.,Economic Return of Clinical Trials Performed Under the Pediatric Exclusivity Program[J].297 JAMA 480,487(2007).
[9] Genebieve Michaux, EU Orphan Regulation - Ten Years of Application[J]. 65 FOOD DRUG L.J. 639, 657 (2010).
[10] David Duffield Rohde, The Orphan Drug Act: An Engine for Innovation? At What Cost? [J].55 FOOD DRUG L.J. 125 , 131 (2000).
[11] 李晓禾、高菲.怪病也得有药医[N].南方周末2010-11-21(4).
[12] Saad Abughanm, The Protection of Pharmaceutical Patents and Data under TRIPS and US-Jordan FTA: Exploring the Limits of Obligations and Flexibilities: A Study of the Impacts on the Pharmaceutical Sector in Jordan[D]. Toronto: University of Toronto, 2012, at 332-333.
[13] Cynthia M. Ho, Access to Medicine in The Global Economy: International Agreements on Patents and Related Rights [M].New York: Oxford University Press, 2011.
[14] Gerrit M. Beckhaus, A New Prescription to Balance Secrecy and Disclosure in Drug-Approval Processes[J]. 46 MICH. J.L.REFORM. 135,137 (2012).
[15] Christopher J. Kochevar, Reforming Judicial Review of Bioequivalence Determinations[J]. 87 N.Y.L.REV. 2040 (2012).
[16] Jason Kanter &Robin Feldman, Understanding and Incentivizing Biosimilars[J].64 HASTINGS L.J. 101 (2012).
