Meta-analysis 在多种组学领域的应用
2014-06-15韩明飞朱云平
韩明飞,朱云平
Meta-analysis 在多种组学领域的应用
韩明飞,朱云平
军事医学科学院放射与辐射医学研究所 蛋白质组学国家重点实验室 国家蛋白质科学中心 (北京) 北京蛋白质组研究中心 蛋白质药物国家工程研究中心,北京 102206
Meta-analysis作为一种整合多特征、多数据的统计方法,上世纪90年代被引入生命科学领域。随着高通量测序技术的快速发展,以基因组学、转录组学和蛋白质组学为核心的生命组学逐渐成为生命科学研究的新热点。海量数据的快速产出推动了组学研究的发展,也引发了数据规模过大、难以系统整合等问题。针对上述情况,meta-analysis被广泛地应用于分析各组学数据,方法也不断得到改进。本文系统总结了有代表性的meta-analysis方法,考察了目前meta-analysis在多个组学领域的应用现状,最后讨论了meta-analysis尚待解决的问题并展望未来的发展方向。
meta-analysis,基因组学,转录组学,蛋白质组学
Meta-analysis的中文名称为“元分析”或“荟萃分析”,最早由Glass在1976年提出[1]。Meta即“more comprehensive”,表示更加全面之意。其分析对象是现有的研究成果,定义为对先前研究的综合评价和定量合并。Meta-analysis最早用于心理、教育等领域,上世纪90年代开始在自然科学领域盛行。时至今日,meta-analysis已经广泛用于生命科学的多个领域,并发展出一系列整合各类数据的方法。
随着高通量技术的快速发展,以基因组、转录组和蛋白质组为核心的生命组学[2]数据大量产出。大规模组学数据库,如Gene Expression Omnibus[3]、ArrayExpress[4]、PeptideAtlas[5]、PRIDE[6]、Encode[7]等,开始进入人们的视野。要深入完整地解开隐藏在大量实验数据中的生物学奥秘,高效的数据整合和分析方法必不可少,meta-analysis就是其中的典型代表。如今,meta-analysis已经被公认为科学有效的数据整合方法,广泛用于各类组学研究。在基因组学领域,meta-analysis主要被用于基因组关联分析(Genome wide association studies,GWAS);在转录组学领域,meta-analysis被广泛用于分析基因芯片数据;在蛋白质组学领域,meta-analysis已经开始被用来整合双向凝胶电泳图谱和质谱数据。
我们在PubMed中以相应关键词搜索了应用于不同组学的meta-analysis研究 (截止到2014年1月10日),共搜集了2 032篇论文。经过初步筛选,与基因组关联分析相关的研究 (基因组学) 有1 092篇,与基因芯片相关的研究(转录组学) 有857篇,与双向电泳、质谱和蛋白数据库 (蛋白质组学) 相关的研究有83篇。图1展示了meta-analysis在3种组学领域相关研究的论文数量以及近十年的发展情况,其中,基因组学和转录组学占主导地位,转录组学的相关研究起步最早,至今一直稳步上升,基因组学相关研究发展最快,2009年已经超越转录组并一直保持强劲的发展势头;蛋白质组学相关的研究起步较晚,正处于逐渐积累的时期。

图1 2005–2013年Pubmed历年收录的meta-analysis在三种组学领域的相关研究论文数Fig. 1 The number of studies about meta-analysis in different areas published in Pubmed from 2005 to 2013.
本文首先介绍了meta-analysis有代表性的算法及相应改进,指出每种方法的优势与不足。之后结合基因组学、转录组学和蛋白质组学的技术特点,考察了meta-analysis在生命组学各领域的应用情况,在此基础上总结现有的meta-analysis分析工具。最后结合自身研究指出meta-analysis存在的问题与相应的解决方案,进而探讨未来meta-analysis的发展方向。
1 Meta-analysis的代表性方法
Ramasamy等在总结大量研究的基础上提出了基因芯片数据meta分析的7个基本步骤[8],其中有些步骤涉及到基因芯片的特异性处理,通过进一步总结,我们归纳出具有普遍性的meta-analysis基本流程:数据收集,数据预处理,特定统计量整合以及结果分析、展示和解释。其中,“特定统计量整合”是meta-analysis最关键的步骤,我们对现有的meta-analysis相关研究进行了系统总结和分类,归纳出4种方法:基于P-value的方法、基于排序 (Rank) 的方法、基于效果量 (Effect size) 的方法以及基于计数的方法。本节将分别介绍4类方法的原理、功能及一些代表性的改进。
1.1 基于P-value的方法
P-value是由统计学检验 (t检验,卡方检验等) 得到的表征某条目 (基因、蛋白质等) 在两个样本中差异显著性的统计量,P值越小代表差异越显著。整合P-value的meta-analysis方法主要用于整合多数据集鉴定差异基因或蛋白质,其原理是综合某条目在所有数据集的P-value大小计算它的综合打分S,以此表征其整合后的差异程度。
计算综合打分的方法有很多。Fisher方法[9]是最经典的方法之一,它取所有P-value的负对数加和作为打分S越大代表差异表达的概率越高。另一种经典方法是Stouffer方法[10],打分P取P-value的标准正态分布的反函数加和。在以上两种方法的基础上,研究人员进行了许多改进,Li和Tseng为每个P-value引入权值0或1,计算所有可能的打分,取其中的最大值作为综合打分,命名为自动调整的加权Fisher方法[11]。此方法比传统方法具有更高的灵敏度,但数据集较多时计算量也大大增加。另一种引入权值的改进是Whitlock提出的加权Z-score方法[12],它根据数据量的大小为不同数据集设置权值,计算加权打分Z。为检验其整合效果,Whitlock使用加权Z-score方法与经典Fisher方法整合同一批数据,发现两个结果具有相似的第一类错误率,但加权Z-score方法的结果表现出较低的第二类错误率以及与标准结果更高的相关性,证明了其更好的整合效能。
1.2 基于排序的方法
尽管整合P-value的方法已被普遍接受,但离群的极小P-value往往在计算综合打分时占据主导地位,导致某基因或蛋白质仅仅因为在某一个数据集中P-value极小而得到一个较高打分,最终被判断为差异。针对这种情况,人们提出了基于排序的方法,它可以有效地降低离群极值的影响,更适于整合质量不一的数据。基于排序的方法将各数据集中的条目根据特定规则 (P-value/Fold-Change) 排序,以位置编号表征其显著程度。其原理与基于P-value的方法相似,仅仅在计算综合打分时用条目的位置编号代替P-value。
Hong等开发了基于排序的meta-analysis工具“RankProd”[13],之后又比较各种meta-analysis方法,认为基于排序的方法相比P-value表现出更好的统计效果[14]。Amess等在经典排序方法的基础上大胆改进,针对蛋白质数据定量准确性较低的情况,提出了ψ-ranking和π-ranking3种表征显著性的新标准,以欧几里德距离作为综合打分,用以整合蛋白质数据。证明3种方法克服了仅依靠Fold-Change作为唯一排序标准的弊端,欧几里德距离的使用也大大降低了整合结果的假阳性[15]。
1.3 基于效果量的方法
基于效果量的方法是最早提出的meta-analysis方法之一,Glass在1976年命名meta-analysis的同时就提出了效果量的概念。概括来说,效果量是描述不同数据间差异大小的指标,例如对于多组不同时期的癌症表达谱,基因表达量的差异就可以作为一个效果量指标,将它标准化后可以衡量生长时间对癌症发展的影响。
基于效果量的meta-analysis方法除了可用于整合多组数据鉴定差异基因或蛋白质,还可以考察不同数据间的异质性。其原理是借助特定的效应模型来拟合多组数据间的差异 (效果量),从而估算效果量大小。固定效应模型(Fixed effects models,FEM) 和随机效应模型(Random effects models,REM) 是两种最常用的拟合效果量的模型,固定效应模型的效果量包括表达量和抽样误差,随机效应模型进一步考虑了不同研究间的差异因此固定效应模型只能用于相同实验条件下的数据,而随机效应模型可用于来源于不同实验的独立数据[16]。
基于效果量的方法是几类meta-analysis方法中功能最强大的。Nakaoka等对用于基因组关联分析的效果量方法进行了总结,介绍了其搜索策略、数据纳入标准和方法具体流程[17]。Choi等尝试将效果量方法用于基因芯片的meta-analysis,详细叙述了鉴定差异基因的具体步骤和算法原理[16]。
1.4 基于计数的方法
基于计数的方法又称计票法(Vote-counting),以某基因或蛋白条目在所有数据集的显著表达列表中的重复次数表征其差异显著性。对于重复多少次为显著性,一般通过随机化方法估算假发现率(False discover rate, FDR),认为使FDR<0.1的重复次数是符合标准的重复次数,重复这些次的基因即为整合后的差异基因[18]。
计票法是一种定性方法,灵敏度较低,在数据量不足时很难作出判断,因此只适用于规模较大的数据。Rhodes等针对大规模数据整合对计票方法进行了改进,并用这种方法整合了包含多于3 700组样本的40个癌症基因芯片数据,取得了不错的效果[18]。计票方法尽管灵敏度有限,却是目前整合大规模数据唯一有效的办法。
2 Meta-analysis在生命组学领域的应用
集多种组学之大成的“生命组学”研究模式已初现端倪[2],迅速积累的各组学数据对高效的数据整合方法提出了更高要求。如今,meta-analysis不仅被广泛用于基因组学、转录组学和蛋白质组学研究,还延伸到组学间的整合分析。
2.1 Meta-analysis在基因组学中的应用
基因组学主要通过研究不同个体基因组的相同和差异探索基因的功能,是最早被提出的组学概念。基因组关联分析是meta-analysis方法应用于基因组学研究的典型代表。基因组关联分析是通过检测特定物种不同个体间的基因序列差异,分析单核苷酸多态性 (Single nucleotide polymorphisms, SNPs) 的方法[19]。检测差异信号往往需要大量样本,单一的基因组关联分析很难得到准确的结果,使用meta-analysis方法整合多个独立研究可以有效降低误报、提高统计能力。Meta-analysis在基因组关联分析中的应用分为两类:第一类是全基因组关联分析,目的是在一个物种全基因组范围内研究单核苷酸多态性;第二类是目标位点分析,有针对性地研究某一基因位点的复制情况[19]。
几乎目前所有对人类多基因疾病的遗传学认识都来自于借助meta-analysis方法的基因组关联分析[20],meta-analysis已成为一种发现疾病和表型新基因位点的普遍方法,用于更大规模样本的meta-analysis还将继续开展,揭示更多基因组层面的生命奥秘[21]。
2.2 Meta-analysis在转录组学中的应用
转录过程是基因表达的第一步,也是基因表达调控的关键环节,转录组学是从转录水平研究基因表达情况的学科。基因芯片是转录组学最重要的研究手段,利用meta-analysis整合芯片数据不仅大大提高了鉴定差异基因的准确率,还衍生出整合通路[22]、整合网络[23]等一系列后续研究。
Meta-analysis整合基因芯片可以实现鉴定差异基因、网络和基因共表达分析[24-27]、预测分析[28]、评估芯片的相似性和差异性[29]等功能。其中鉴定差异基因应用最为广泛,除了两个样本的差异,也有针对连续、多级变量等多个样本间差异展开的研究[30]。此外,meta-analysis整合多组基因芯片与后续生物学研究的结合也越来越紧密。例如,Shen等开发的工具MAPE[31]将meta-analysis和通路富集分析巧妙地结合在一起。通过鉴定差异基因与通路 (Gene Ontology,KEGG[32]等数据库) 的相关性,判断一个已知的生物过程是否在差异基因列表中富集。Yang等收集了6个物种的基因芯片数据用于鉴定它们全部基因的编码蛋白,与不同条件下的基因表达数据整合分析,研究不同物种及不同条件下的蛋白结构域特点[33]。
基因芯片凭借其方便高效的优势,近年被广泛用于研究疾病或生物处理前后的基因表达差异。然而受到技术限制和实验偶然因素的影响,单次实验的准确性还有待提高。用meta-analysis方法整合多基因芯片可以有效降低各种偶然因素的影响,揭示多次实验一致的表达规律。
2.3 Meta-analysis在蛋白质组学中的应用
随着人类基因组计划的实施和推进,生命科学已进入了后基因组时代,蛋白质组学不仅是生命科学研究进入后基因组时代的里程碑,也是后基因组时代生命科学研究的核心内容之一。蛋白质鉴定是蛋白质组学最重要的研究内容之一,凝胶电泳和质谱是两大关键技术,双向电泳图谱的meta-analysis出现较早,近年随着质谱技术的发展,质谱数据的meta-analysis也逐渐展开。
整合双向电泳图谱有时可以揭示关于生物过程的隐藏信息。Natale等通过整合两个电泳图谱研究帕金森症疾病相关蛋白DJ-1[34]。多样本整合也得以应用,Rosenberg等通过整合73个肿瘤样本的电泳图谱中2 121个点研究前列腺和结肠肿瘤的蛋白表达[35]。在众多的高通量技术中,质谱被认为是一种同时具备高特异性和高灵敏度且得到了广泛应用的普适性方法。整合质谱数据有整合原始数据和整合处理后的蛋白质列表两种方式。原始数据较为理想,但相对不容易获得,相反经过预处理的蛋白质列表来源就广泛得多。整合原始数据时一般对这些数据进行统一搜库,以保证得到标准化的蛋白质ID,然后整合各数据集内蛋白质的FDR[36]。整合蛋白质列表时现有的meta-analysis方法基本都可以使用,Amess等改进了基于排序的meta-analysis方法,用改进方法整合多组蛋白质列表,取得了很好的效果[15]。除鉴定蛋白质外,meta-analysis还被用于分析蛋白质的丰度和结构特点。Zhong等整合分析了6个物种的蛋白丰度数据,考察了不同物种间以及不同结构域下蛋白丰度的分布模式[37]。
蛋白质是生理功能的执行者,是生命现象的直接体现者,对蛋白质结构和功能的研究将直接阐明生命在生理或病理条件下的变化机制。蛋白质的可变性和多样性等特殊性质导致了蛋白质研究技术远远比核酸技术要复杂,meta-analysis整合多次实验可以有效弥补蛋白质技术目前在准确性上的不足,推动蛋白质研究迅速发展。
2.4 Meta-analysis在多组学整合中的应用
早期组学间整合的主要目的是通过比较两组学数据,评估两类数据的相似性,其中最多的是转录组和蛋白质组的比较分析,较普遍的比较方法有Nie等提出的零堆积泊松模型(Zero-inflated Poisson model)[38]和Kislinger等建立的贝叶斯网络[39]。后来组学整合的目的逐渐拓展为探索不同组学的内在关联以揭示生物系统的作用机制。例如,Lv等通过整合转录组和毒理基因组数据识别癌症先导化合物[40],Liu等通过整合转录组和蛋白质组二维凝胶电泳数据鉴定先兆子痫的生物标志物[41]。最近,Sass等提出了一种基于贝叶斯模型方法——多层次本体分析算法 (Multi-level Ontology Analysis, MONA),可用于综合分析多组学数据并评估其生物学意义[42]。图2展示了PubMed历年收录的多组学整合相关研究论文数。图中可见,组学间整合研究总体呈增长趋势,2013年更是呈现大幅度增长。随着组学研究的深入,我们有理由预测这一领域将在未来占据重要 地位。
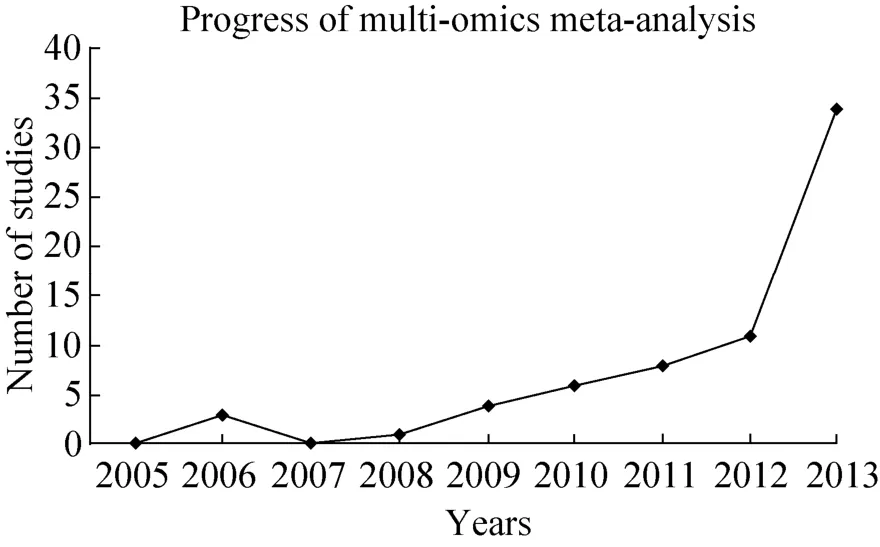
图2 近十年Pubmed历年收录的多组学整合相关论文Fig. 2 The number of studies about integration of multi-omics published in Pubmed for this decade.
3 Meta-analysis代表性工具
前文讨论了meta-analysis在基因组、转录组、蛋白质组以及多组学整合4个领域的应用。针对不同的研究目的,目前已经开发了一些具备meta-analysis功能的工具。与一般组学数据分析工具以单一数据为基本单位不同,meta-analysis工具从不同来源的数据入手,以整合分析为基本途径,一般实现一种或几种meta-analysis算法,最终实现鉴定、预测以及可视化等功能。
在基因组学领域,meta-analysis相关工具发展较成熟,其中使用最广泛的是METAL[43],它实现了加权Z-score和基于效应量的整合方法;PLINK实现了FEM和REM方法,是一个免费、开源的基因组关联分析工具[44];Comprehensive Meta-analysis (CMA)是一个商业软件,它具有电子表格形式的界面并可以绘制森林图[45]。此外,基因组学领域的meta-analysis工具还包括“MAGENTA”[46](利用meta-analysis实现基因集富集分析)、Metafor[47](实现多种基于效应量的方法并提供多种图形展示)、Synthesis-view[48](整合多组研究并可视化结果)、IGG3[49](整合GWAS原始数据) 以及GWAMA[50](实现FEM和REM方法)。
在转录组学领域,科研人员同样开发了一些针对基因芯片数据的meta-analysis工具和软件包,其中发展较成熟的包括:GeneMeta是Bioconductor环境下的软件包,实现了FEM和REM方法, metaMA是R语言环境下的软件包,实现了REM方法和Stouffer方法,metaArray[51]可以整合多组表达概率 (Probability of expression,POE),而OrderedList[52]、RankProd[13]和RankAggreg[53]三个软件包均实现了基于排序的meta-analysis方法。总体而言,目前整合基因芯片的meta-analysis工具与流行的芯片处理工具 (如SAM、PAM等) 相比还有待改善,大部分缺乏用户友好的界面和使用手册[54],因此优化算法并开发准确好用的工具在转录组学领域仍具有重大价值。
蛋白质组学起步较晚,数据积累有限,且蛋白质组数据具有特殊性,例如重复实验数目不足,不同蛋白的丰度无法比较等[55],因此单纯整合蛋白质实验数据的工具或软件包还很有限,更多的是利用meta-analysis算法整合蛋白质数据实现特定功能,例如,PTMeta[56]利用meta-analysis整合质谱数据不同修饰条件下的搜库结果,用于鉴定修饰肽段。Morphinome[57]是一个蛋白数据库,它利用meta-analysis整合了15组神经系统受吗啡影响后的蛋白表达数据,可以进行吗啡影响的预测;motif-x和scan-x[58]利用meta-analysis整合了不同物种的翻译后修饰信息,可以用于磷酸化与乙酰化位点预测。蛋白质组学领域相关工具的缺乏也为其开发提出了迫切需求,要更好地分析不断积累的蛋白质组学数据,meta-analysis将是重要的研究内容。
表1列举了meta-analysis在不同研究领域有代表性的工具及其功能介绍。
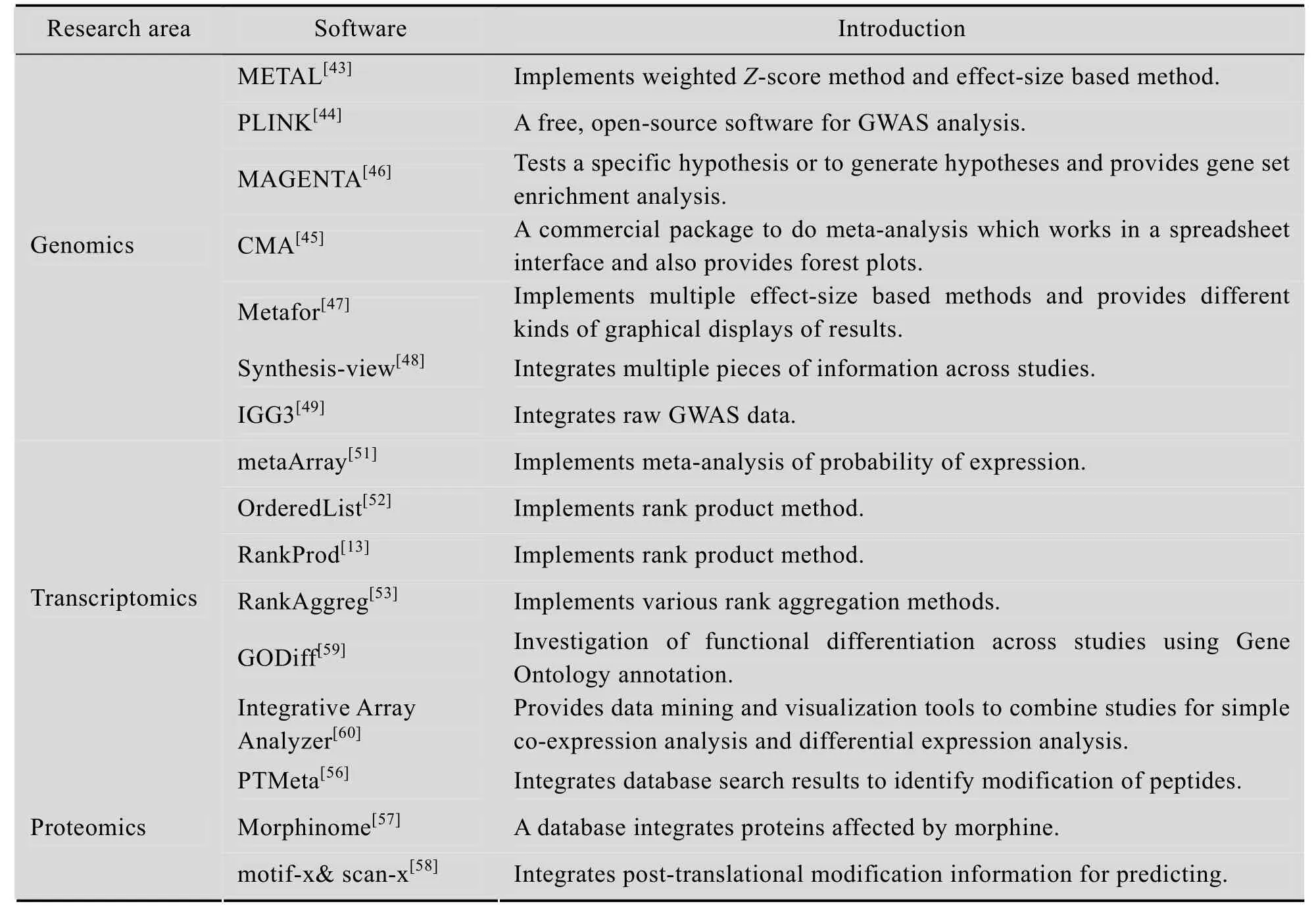
表1 Meta-analysis工具总结Table 1 Summary of meta-analysis tools
4 小结与展望
随着多组学数据的不断积累,有效地处理数据并整合分析将变得越来越重要。与此同时,我们也清醒地认识到meta-analysis尚存在一些待解决的问题。在实验技术层面,数据质量不统一特别是离群数据的存在在一定程度上影响了整合结果,因此在整合数据时不能一味地追求数据的完整性,而应该建立有效的数据评估机制,除了在检索数据库时选择准确有效的关键词外,还要根据数据的样本量、实验平台等条件进行主观筛选,并在此基础上通过求相关系数和聚类等方式剔除离群数据。Ramasamy等提出整合各数据的“可重复基因” (Reproducible genes)[8]也可以有效缓解数据质量不一的问题。在方法推广层面,目前的meta-analysis工具实用性还很有限,大部分只局限于特定研究,缺乏用户友好的界面和使用手册。这对开发方便实用的软件提出了迫切需求,特别是建立起集成各种方法的通用的meta-analysis整合工作流,不仅利于meta-analysis推广到更多领域,还能促进现有方法的改进和新方法的出现。
Meta-analysis正在生命科学研究中扮演着重要角色。无论是发展较成熟的基因组学、转录组学,还是迅速崛起的蛋白质组学,meta-analysis都得到广泛应用并发挥了巨大价值。从社会学到生物学,从基因组学到蛋白质组学,其发展经历了一个不断推广到新领域、应用于新数据的过程。我们有理由预测,未来要建立集所有组学的“生命组学”研究模式,meta-analysis将被推广到更多新领域,体现越来越大的价值。
REFERENCES
[1] Smith ML, Glass GV. Meta-analysis of psychotherapy outcome studies. Am Psychol, 1977, 32(9): 752–760.
[2] He F. Lifeomics leads the age of grand discoveries. Sci China Life Sci, 2013, 56(3): 201–212.
[3] Bhargava A, Clabaugh I, To JP, et al. Identification of cytokinin-responsive genes using microarray meta-analysis and RNA-Seq in Arabidopsis. Plant Physiol, 2013, 162(1): 272–294.
[4] Parkinson H, Kapushesky M, Shojatalab M, et al. ArrayExpress--a public database of microarray experiments and gene expression profiles. Nucleic Acids Res, 2007, 35(Database issue): D747–750.
[5] Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep, 2008, 9(5): 429–434.
[6] Martens L, Hermjakob H, Jones P, et al. PRIDE: the proteomics identifications database. Proteomics, 2005, 5(13): 3537–3545.
[7] Maher B. ENCODE: The human encyclopaedia. Nature, 2012, 489(7414): 46–48.
[8] Ramasamy A, Mondry A, Holmes CC, et al. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med, 2008, 5(9): e184.
[9] Rhodes DR, Barrette TR, Rubin MA, et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res, 2002, 62(15): 4427–4433.
[10] Stouffer SA. A study of attitudes. Sci Am, 1949, 180(5): 11–15.
[11] Li J, Tseng GC. An adaptively weighted statistic for detecting differential gene expression when combining multiple transcriptomic studies. Ann App Stat, 2011, 5(2A): 994–1019.
[12] Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J Evol Biol, 2005, 18(5): 1368–1373.
[13] Hong F, Breitling R, McEntee CW, et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics, 2006, 22(22): 2825–2827.
[14] Hong F, Breitling R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics, 2008, 24(3): 374–382.
[15] Amess B, Kluge W, Schwarz E, et al. Application of meta-analysis methods for identifying proteomic expression level differences. Proteomics, 2013, 13(14): 2072–2076.
[16] Choi JK, Yu U, Kim S, et al. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics, 2003, 19(Suppl 1): i84–90.
[17] Nakaoka H, Inoue I. Meta-analysis of genetic association studies: methodologies, between-study heterogeneity and winner's curse. J Hum Genet, 2009, 54(11): 615–623.
[18] Rhodes DR, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA, 2004, 101(25): 9309–9314.
[19] Begum F, Ghosh D, Tseng GC, et al. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res, 2012, 40(9): 3777–3784.
[20] Panagiotou OA, Willer CJ, Hirschhorn JN, et al. The power of meta-analysis in genome-wideassociation studies. Annu Rev Genomics Hum Genet, 2013, 14: 441–465.
[21] Thompson JR, Attia J, Minelli C. The meta-analysis of genome-wide association studies. Brief Bioinform, 2011, 12(3): 259–269.
[22] Kaever A, Landesfeind M, Feussner K, et al. Meta-analysis of pathway enrichment: combining independent and dependent omics data sets. PLoS ONE, 2014, 9(2): e89297.
[23] Wang K, Narayanan M, Zhong H, et al. Meta-analysis of inter-species liver co-expression networks elucidates traits associated with common human diseases. PLoS Comput Biol, 2009, 5(12): e1000616.
[24] Mabbott NA, Kenneth Baillie J, Hume DA, et al. Meta-analysis of lineage-specific gene expression signatures in mouse leukocyte populations. Immunobiology, 2010, 215(9/10): 724–736.
[25] Carrera J, Rodrigo G, Jaramillo A, et al. Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol, 2009, 10(9): R96.
[26] Jupiter D, Chen H, VanBuren V. STARNET 2: a web-based tool for accelerating discovery of gene regulatory networks using microarray co-expression data. BMC Bioinformatics, 2009, 10: 332.
[27] Mehan MR, Nunez-Iglesias J, Kalakrishnan M, et al. An integrative network approach to map the transcriptome to the phenome. J Comput Biol, 2009, 16(8): 1023–1034.
[28] Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst, 2010, 102(7): 464–474.
[29] Nguyen VA, Lio P. Measuring similarity between gene expression profiles: a Bayesian approach. BMC Genomics, 2009, 10(Suppl 3): S14.
[30] Lu S, Li J, Song C, et al. Biomarker detection in the integration of multiple multi-class genomic studies. Bioinformatics, 2010, 26(3): 333–340.
[31] Shen K, Tseng GC. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics, 2010, 26(10): 1316–1323.
[32] Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA, 2005, 102(43): 15545–15550.
[33] Yang D, Zhong F, Li D, et al. General trends in the utilization of structural factors contributing to biological complexity. Mol Biol Evol, 2012, 29(8): 1957–1968.
[34] Natale M, Bonino D, Consoli P, et al. A meta-analysis of two-dimensional electrophoresis pattern of the Parkinson's disease-related protein DJ-1. Bioinformatics, 2010, 26(7): 946–952.
[35] Rosenberg LH, Franzen B, Auer G, et al. Multivariate meta-analysis of proteomics data from human prostate and colon tumours. BMC Bioinformatics, 2010, 11: 468.
[36] Higdon R, Haynes W, Kolker E. Meta-analysis for protein identification: a case study on yeast data. OMICS, 2010, 14(3): 309–314.
[37] Zhong F, Yang D, Hao Y, et al. Regular patterns for proteome-wide distribution of protein abundance across species. PLoS ONE, 2012, 7(3): e32423.
[38] Nie L, Wu G, Brockman FJ, et al. Integrated analysis of transcriptomic and proteomic data of Desulfovibrio vulgaris: zero-inflated Poisson regression models to predict abundance of undetected proteins. Bioinformatics, 2006, 22(13): 1641–1647.
[39] Kislinger T, Cox B, Kannan A, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell, 2006, 125(1): 173–186.
[40] Lv S, Xu Y, Chen X, et al. Prioritizing cancer therapeutic small molecules by integrating multiple OMICS datasets. OMICS, 2012, 16(10): 552–559.
[41] Liu LY, Yang T, Ji J, et al. Integrating multiple'omics' analyses identifies serological protein biomarkers for preeclampsia. BMC Med, 2013, 11(1): 236.
[42] Sass S, Buettner F, Mueller NS, et al. A modular framework for gene set analysis integrating multilevel omics data. Nucleic Acids Res, 2013, 41(21): 9622–9633.
[43] Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 2010, 26(17): 2190–2191.
[44] Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007, 81(3): 559–575.
[45] Qu HQ, Bradfield JP, Li Q, et al. In silico replication of the genome-wide association results of the Type 1 Diabetes Genetics Consortium. Hum Mol Genet, 2010, 19(12): 2534–2538.
[46] Segre AV, Groop L, Mootha VK, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet, 2010, 6(8): 264–268.
[47] Viechtbauer W. Conducting Meta-Analyses in R with the metafor Package. J Stat Softw, 2010, 36(3): 1–48.
[48] Pendergrass SA, Dudek SM, Crawford DC, et al. Synthesis-View: visualization and interpretation of SNP association results for multi-cohort, multi-phenotype data and meta-analysis. BioData Min, 2010, 3: 10.
[49] Li MX, Jiang L, Kao PY, et al. IGG3: a tool to rapidly integrate large genotype datasets for whole-genome imputation and individual-level meta-analysis. Bioinformatics, 2009, 25(11): 1449–1450.
[50] Magi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics, 2010, 11: 288.
[51] Choi H, Shen R, Chinnaiyan AM, et al. A latent variable approach for meta-analysis of gene expression data from multiple microarray experiments. BMC Bioinformatics, 2007, 8: 364.
[52] Lottaz C, Yang X, Scheid S, et al. OrderedList--a bioconductor package for detecting similarity in ordered gene lists. Bioinformatics, 2006, 22(18): 2315–2316.
[53] Pihur V, Datta S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinformatics, 2009, 10: 62.
[54] Tseng GC, Ghosh D, Feingold E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res, 2012, 40(9): 3785–3799.
[55] Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol, 2008, 26(12): 1367–1372.
[56] Nahnsen S, Sachsenberg T, Kohlbacher O. PTMeta: increasing identification rates of modified peptides using modification prescanning and meta-analysis. Proteomics, 2013, 13(6): 1042–1051.
[57] Bodzon-Kulakowska A, Kulakowski K, Drabik A, et al. Morphinome--a meta-analysis applied to proteomics studies in morphine dependence. Proteomics, 2011, 11(1): 5–21.
[58] Schwartz D, Chou MF, Church GM. Predicting protein post-translational modifications using meta-analysis of proteome scale data sets. Mol Cell Proteomics, 2009, 8(2): 365–379.
[59] Chen Z, Wang W, Ling XB, et al. GO-Diff: mining functional differentiation between EST-based transcriptomes. BMC Bioinformatics, 2006, 7: 72. [60] Pan F, Kamath K, Zhang K, et al. Integrative Array Analyzer: a software package for analysis of cross-platform and cross-species microarray data. Bioinformatics, 2006, 22(13): 1665–1667.
(本文责编 郝丽芳)
Applications of meta-analysis in multi-omics
Mingfei Han, and Yunping Zhu
Beijing Proteome Research Center, State Key Lab oratory of Proteomics, National Engineering Res earch Center for Protein Drugs, National Center for Protein Sciences Beijing, Beijing Institute of Radiation Medicine, Beijing 102206, China
As a statistical method integrating multi-features and multi-data, meta-analysis was introduced to the field of life science in the 1990s. With the rapid advances in high-throughput technologies, life omics, the core of which are genomics, transcriptomics and proteomics, is becoming the new hot spot of life science. Although the fast output of massivedata has promoted the development of omics study, it results in excessive data that are difficult to integrate systematically. In this case, meta-analysis is frequently applied to analyze different types of data and is improved continuously. Here, we first summarize the representative meta-analysis methods systematically, and then study the current applications of meta-analysis in various omics fields, finally we discuss the still-existing problems and the future development of meta-analysis.
meta-analysis, genomics, transcriptomics, proteomics
April 2, 2014; Accepted: May 13, 2014
Yunping Zhu. Tel/Fax: +86-10-80705225; E-mail: zhuyunping@gmail.com
韩明飞, 朱云平. Meta-analysis在多种组学领域的应用. 生物工程学报, 2014, 30(7): 1094−1104.
Han MF, Zhu YP. Applications of meta-analysis in multi-omics. Chin J Biotech, 2014, 30(7): 1094−1104.
Supported by: National Basic Research Program of China (973 Program) (Nos. 2011CB910600, 2010CB912700, 2013CB911200), National High Technology Research and Development Program of China (863 Program) (Nos. 2012AA020409, 2012AA020201), National Natural Science Foundation of China (Nos. 21105121, 21275160), National Natural Science Foundation of Beijing (No. 5122013).
国家重点基础研究发展计划 (973计划) (Nos. 2011CB910600,2010CB912700,2013CB911200),国家高技术研究发展计划 (863计划) (Nos. 2012AA020409, 2012AA020201),国家自然科学基金 (Nos. 21105121, 21275160),北京市自然科学基金 (No. 5122013)资助。
