原核生物蛋白质基因组学研究进展
2014-06-15张成普徐平朱云平
张成普,徐平,朱云平
原核生物蛋白质基因组学研究进展
张成普,徐平,朱云平
军事医学科学院放射与辐射医学研究所 蛋白质组学国家重点实验室 国家蛋白质科学中心 (北京) 北京蛋白质组研究中心 蛋白质药物国家工程研究中心,北京 102206
随着基因组测序技术的不断发展,大量微生物基因组序列可以在短时间内得以准确鉴定。为了进一步探究基因组的结构与功能,基于序列特征与同源特征的基因组注释算法广泛应用于新测序物种。然而受基因组测序质量以及算法本身准确性偏低等问题的影响,现有的基因组注释存在着相当比例的假基因以及注释错误,尤其是蛋白质N端的注释错误。为了弥补基因组注释的不足,以基因芯片或RNA-seq为核心的转录组测序技术和以串联质谱为核心的蛋白质组测序技术可以高通量地对基因的转录和翻译产物进行精确测定,进而实现预测基因结构的实验验证。然而,原核生物细胞中存在的大量非编码RNA给转录组测序技术引入了污染数据,限制了其对基因组注释的应用。相对而言,以串联质谱技术为核心的蛋白质组学测序可以在短时间内鉴定到生物体内大量的蛋白质,实现注释基因的验证甚至校准。已成为基因组注释和重注释的重要依据,并因而衍生了“蛋白质基因组学”的新研究方向。文中首先介绍传统的基于序列预测和同源比对的基因组注释算法,指出其中存在的不足。在此基础上,结合转录组学与蛋白质组学的技术特点,分析蛋白质组学对于原核生物基因组注释的优势,总结现阶段大规模蛋白质基因组学研究的进展情况。最后从信息学角度指出当前蛋白质组数据进行基因组重注释存在的问题与相应的解决方案,进而探讨未来蛋白质基因组学的发展方向。
蛋白质基因组学,原核生物,基因组注释,质谱
自1995年首个原核生物实现全基因组测序至今[1],基因组测序技术的快速发展已经实现对古生菌 (Archaea)、 细菌 (Bacteria) 以及真核生物 (Eukaryotes) 等3界中11 176个物种序列的精确测定,其中原核生物4 572个,占到了总数的40.9% (基于NCBI 2013年12月统计结果)。为了充分解析基因组的结构和功能,基因组注释得到了快速推广[2-4]。相对于真核生物而言,原核生物基因组基因数目较少,大部分序列属于编码基因。依托序列预测和同源比对,大量原核生物基因组得以批量化注释。然而受基因组测序质量影响以及缺乏合适的校正评估机制,原核生物基因组注释存在相当比例的假基因和注释错误,尤其是蛋白质N端的注释错误,给充分研究相应物种的生理学机制带来了困难[5-6]。为了解决这一问题,多种新兴技术策略开始采用实验数据集对基因组基因注释进行校正。最为典型的是以RNA-seq或基因芯片为核心的转录组测序技术[7]和以串联质谱技术为核心的蛋白质组测序技术[8]。其中基于串联质谱的蛋白质组学技术研究可通过大规模、高通量地测定基因表达终产物蛋白质的序列,是有别于核酸测序的相对独立的技术手段,不仅可以验证已注释基因,还可对已注释基因的结构进行修正和鉴定未被传统基因组注释算法注释的基因,发现新的基因结构特征。这对于基因组学本身的发展和研究相应物种的生物学特性具有十分重要的意义。
从转录组与蛋白质组的比较来看,原核生物细胞中存在有大量的非编码RNA,这给转录组测序技术引入了较多的污染数据,限制了其对原核生物基因组注释的应用[9-10]。相对而言,以串联质谱为核心的蛋白质组学测序技术可以在短时间内鉴定到生物体或细胞内大量的蛋白质,实现对于基因表达最直接的验证,已成为基因组注释的重要依据之一,“蛋白质基因组学”应运而生[11]。如今,快速发展的质谱技术令人们可以对原核生物的基因组有着较高的鉴定蛋白质比例和鉴定肽段覆盖度,实现基因组注释的高通量、规模化重注释。利用原核生物基因组相对简单的特点,通过数据库搜索的方式检索六阅读框翻译的基因组数据库 (Six-reading Frame translated Genome Database) 并与已注释蛋白质进行比对,可以快速准确检索出注释数据库存在和不存在的肽段及其对应的基因,在验证已有注释基因的同时实现对基因组注释的修正。
本文首先介绍传统基于序列预测和同源比对的基因组注释算法,指出其中存在的不足。之后结合转录组学与蛋白质组学的技术特点,分析蛋白质组学对原核生物基因组重注释的优势,总结现阶段大规模蛋白质基因组学研究的进展情况。最后从信息学角度指出当前蛋白质组数据进行基因组重注释存在的问题与相应的解决方案,进而探讨未来蛋白质基因组学的发展方向。
1 原核生物基因组注释研究背景
早在1995年嗜血流感菌Haemophilus influenza被作为首个物种实现基因组全测序之前,研究者就开始思考探索相应的基因注释方案[4]。利用基因组序列特征,一系列基因组注释软件被广泛地应用于测序结果的基因注释,例如Glimmer[12]、GenemarkHMM[13]以及Easygene[2]等。不同于人或者小鼠等研究广泛的真核生物,数目众多的原核生物对应的基因组注释很少有充分的实验验证。大多数原核生物在实现基因组测序之后,基因结构的确定都会使用自动化注释软件实现,功能注释则采用同源比对实现[2]。随着基因组数目的不断增加,自动化注释流程在准确性上的不足逐渐显现。已有研究表明,目前已注释的原核生物基因组中,有一半以上的基因结构存在注释不准确的现象[2,10],其中蛋白质N端的注释错误尤为明显,即便是对于研究广泛的大肠杆菌也是如此[14-15]。造成这种现象的原因主要有3个方面:一是基因组测序中存在的错误导致注释的不准确;二是通用性的序列预测特征不一定适用于各个物种,尤其是对于一些存在于短阅读框区的基因,很难实现准确预测;三是对原核生物的N端缺少合适的特征,导致N端具有较高的注释错误率。采用序列比对的方式进行功能注释仅适用于物种间的直系同源基因,对于一些物种特有基因则无法进行功能注释,这也是原核生物中存在大量“假定蛋白质”(Hypothetical protein) 的重要原因。
为了弥补传统基因组注释方法的不足。具有高通量、高基因组覆盖度的转录组测序技术被广泛地应用于基因组注释研究,尤其是真核生物的基因组校正分析[3,16-18]。然而对于原核生物,细胞中存在的大量非编码RNA容易对转录组测序造成污染,导致一些与非编码RNA序列相似的编码RNA难以得到准确的测定[9-10]。此外,由于蛋白质成熟过程中N端降解现象的存在[10,19],直接对细胞中存在的蛋白质进行高通量测定更有利于准确注释基因结构,挖掘相应基因的生物学功能。
2 利用蛋白质组数据对原核生物基因组进行重注释
以高速发展的质谱技术为核心的蛋白质组学已经成为后基因组时代研究的热点之一[8]。鸟枪法策略是目前蛋白质组学领域使用最为广泛的技术路线。该策略首先对分离得到的蛋白质混合物进行酶解成为肽段混合物,之后进行液相色谱分离,最后由串联质谱产出肽段匹配信息。数据库搜索结合蛋白质水平的质控策略是目前分析大规模质谱数据的最主要手段[20-21]。在得到高可信的肽段鉴定列表之后,通过蛋白质装配获得鉴定蛋白质列表,进而确定样品中鉴定得到的表达基因及其相应的匹配肽段。
相对于转录组测序而言,目前蛋白质组学技术的最大问题是基因组鉴定覆盖度偏低。以人类相关研究为例,已有的大规模蛋白质组学数据集的基因组覆盖度一般在50%−60%之间,而转录组数据的基因组覆盖度一般在70%以上[22-23]。造成这种现象的原因主要是蛋白质在时空表达上的差异以及质谱技术的局限[24]。相对于真核生物,原核生物基因组相对较为简单,对应的基因数目也相对较少。单细胞结构以及相对简单的表达调控机制,有利于用蛋白质组学技术对细胞内的所有蛋白质进行高通量测定[25]。
2.1 蛋白质组学数据进行基因组重注释基本原理
利用蛋白质组学数据进行基因组重注释的首要任务是构建六框翻译数据库,即人为地将双链DNA按照三联密码子的排列方式穷举所有的可翻译蛋白质,每两个终止子之间的DNA序列定义为一个“开放阅读框”(Open reading frame, ORF)。原核生物不含内含子,采用六编码阅读框方式直接翻译基因组序列,并与已注释的蛋白质序列库合并进行数据库搜索,找寻相应的鉴定肽段,从而在验证并修正已注释基因结构的同时发现未注释基因。
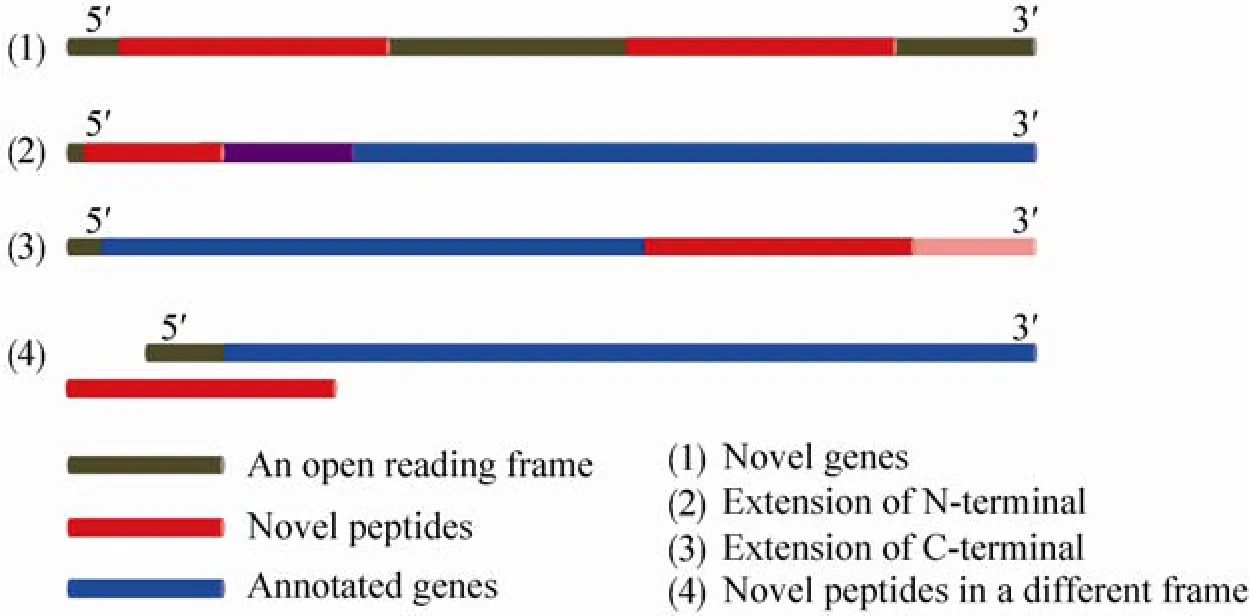
图1 新肽段编码序列与已注释基因染色体位置邻近关系Fig. 1 The relationship between novel peptides and annotated genes on the chromosome.
鉴定得到的高可信肽段会被分成两类:一类是属于已注释基因的肽段,主要用于验证相因基因注释的准确性;另一类是不属于已注释基因的新肽段。根据这些新肽段与已注释基因的邻近关系可以进一步分成4类,如图1所示。第一类是新基因,即新肽段处于一个独立的阅读框中,并且不与已注释基因有过多重叠。第二类是注释基因N端的延伸,要求新肽段与原注释基因在同一个阅读框内。第三类是注释基因C端的延伸,通常对应于终止子测序错误、核苷酸突变或终止密码翻译通读。第四类是注释基因发生了具有阅读框移动的延伸,即新肽段与已注释基因相邻或者部分重叠,但是不处于同一个阅读框中,并且没有充分的证据证明新基因的存在。第四类新肽段的产生可能是由于基因本身翻译后的移码 (Frameshift)[26],但更主要是由于基因组测序错误所导致[27]。
尽管原核生物不需要像真核生物一样在六框翻译数据库搜索时考虑搜索空间过大的问题[28],但仍需要在常规质量控制[29]的基础上对新肽段进行更加严格过滤卡值,以降低新肽段鉴定错误匹配的可能[30]。在鉴定得到新肽段列表之后,需要对其进行质谱证据以外的验证。目前常见的新肽段验证方式主要有3个:一是采用序列特征分析、同源比对以及功能分析等方式在信息学角度进行验证;二是采用RT-PCR[31]对特定的基因表达区域进行验证;三是采用合成肽段的方式确认谱图匹配的准确性或者结合质谱多离子反应监测 (Multiple reaction monitoring,MRM) 和选择反应监测 (Selective reaction monitoring,SRM)[32]技术进行验证。
2.2 蛋白质基因组学发展历史与现状
早在1995年,Yates等已经将蛋白质组数据应用于DNA序列库搜索[33],但并没有系统地对某个物种的基因组注释进行修正。蛋白质基因组学始于2004年Jaffe等对于肺炎支原体的研究[34]。尽管当时技术条件有限,但对于像支原体等规模很小的基因组 (约810 kb) 以及相对简单的细胞结构,他们验证了81%的注释基因,并利用蛋白质组学数据修正了将近10%的已注释基因结构。随着蛋白质组学技术的不断发展,越来越多的人们开始质疑基因组测序及其注释的准确性,并采用以质谱为核心的蛋白质学组技术对基因组注释进行修正。
表1列举了近十年来原核生物蛋白质基因组学的部分研究成果。基因组重注释对于样本制备或数据没有特殊要求,对计算资源的需求也相对较小,原核生物蛋白质基因组学研究已经形成了数据产出→六阅读框翻译数据库搜索→新基因挖掘的流程化模式。研究显示通过蛋白质基因组学鉴定的新基因的数目与数据规模也呈现出一定的正相关性。随着时间的推移,单一的重注释分析已经难以吸引人们的眼球,研究者开始关注于新基因或者已有基因形式修正的生物学意义。比如Baudet等对沙漠奇异球菌Deinococcus deserti的研究专门关注于N端注释的修正[35],而不是全谱的覆盖,因此即使其鉴定基因数目不多,也可以实现对于60个基因N端注释的修正,并验证了若干对生理机制十分重要的非经典起始编码的基因。Gupta等在对奥奈达湖希瓦氏菌Shewanella oneidensis的研究中不仅关注于新基因和N端注释的修正,还对翻译后修饰进行了系统研究,发现并验证了9种高可信体内修饰,成为首个针对翻译后修饰的蛋白质基因组学研究[36]。在定量方面,Chen等对腾冲嗜热杆菌Thermoanaerobacter tengcongensis在不同温度环境下的基因表达状况进行了基于同位素标记 (Isobaric tags for relative and absolute quantitation,iTRAQ) 的定量比较分析[37],找到了高可信的温度相关基因,并用转录组手段进行了验证。尽管该文未进行新基因的挖掘,但提示研究者们不断发展的蛋白质组学定量技术可以和基因组注释的修正相联系,更好地揭示相关物种的生物学特性。
具有较高基因组覆盖度以及鉴定肽段覆盖度的蛋白质组学数据可以对现有的基因组注释进行有效的修正,但却必须建立在该物种基因组测序正确的基础上进行。此外,受环境变化等因素的影响,原核生物功能和进化相对较大的多样性[27]也会令这些物种的基因组发生一定的改变。为了弥补这一点,蛋白质基因组学的研究不再局限于单一物种,开始对多个物种进行比较分析,以期实现交叉验证,进一步提升基因注释修正的规模和准确性。
2.3 比较蛋白质基因组学
在基因组时代,利用物种间亲缘关系进行同源比对获取相应基因的注释信息十分常见。蛋白质组学测序通量的不断提升促使人们得以在蛋白质水平比较不同物种的表达差异。这对于揭示近源微生物表型差异发挥了重要的作用[42]。从注释修正的角度考虑,参考多个物种的基因组序列及其注释情况还有助于校正基因组测序中存在的错误,弥补蛋白质组测序目前还普遍存在的肽段覆盖度不足的难题。比较蛋白质组学研究可以分成两个层面:一是在实验数据层面进行比较;二是在数据分析层面进行比较。

表1 原核生物蛋白质基因组注释主要研究成果Table 1 List of proteogenomic analyses in prokaryotes
在实验数据层面进行比较分析是指对若干近源物种的蛋白质组学数据进行比较分析,在实现基因组重注释的同时还可以在蛋白质层面比较相应物种的表达差异,找寻重要的功能基因。2008年Gupta等首先对希瓦氏菌Shewanella的3个近源菌株进行了比较蛋白质基因组学研究[43]。通过确定3个菌株共有的直系同源基因,提升了这些基因的鉴定肽段覆盖度,降低了单肽段蛋白质比例。在注释修正方面,除了传统的新基因挖掘和N端注释的修正,该工作还发现了12个移码现象以及相当比例的基因组测序错误。此后,对多个近源物种 (一般2−3个) 的基因组进行蛋白质水平的比较分析,成为了蛋白质基因组学研究的新趋势,比如Alexova等对铜绿微囊藻Microcystis aeruginosa的比较分析[44]、Schrimpe-Rutledge等对3种耶尔森氏菌Yersinia菌株的研究[45]以及Zhong等对钩端螺旋体Leptospira interro gans致病基因的分析[46]等。
尽管对多个物种的蛋白质组数据进行比较分析有利于提升鉴定基因的比例并提升基因组重注释的质量,但也提高了实验操作的负担,尤其是只需要对单一物种进行分析的情况。针对这一问题,人们开始在数据层面进行比较蛋白质基因组学分析。即仅对单一物种进行蛋白质组学测序,并以该物种为核心,其他物种为背景进行比较分析,最终只对核心物种的基因组注释进行修正。这种分析策略一般需要引入多种与核心物种近源的背景物种,以提高注释修正的准确性。比如2009年Gallien等对耻垢分枝杆菌Mycobacterium smegmatis的质谱数据进行数据库搜索时,还同时考虑16种分枝杆菌近源菌株[40],有效地提升了N端注释的修正数目。由于使用了较多的背景物种,合理地确定直系同源基因是比较蛋白质基因组学研究的重要问题。目前使用较为广泛算法可以分为两类:一是以PipeAlign[47]和MUSCLE[48]为代表的多重比对算法,优点是多物种比较、准确性高,不足之处是运算速度慢;二是以Remm等使用的方法为代表的两两比对算法[49],可以快速找出两个物种的直系同源基因。
比较蛋白质基因组学可以实现基因组学与蛋白质组学的优势互补,提升基因组重注释修正基因的规模与准确性,并已成为如今原核生物蛋白质基因组研究广泛使用的策略[11]。
3 原核生物蛋白质基因组学研究存在的问题与对策
尽管针对原核生物的蛋白质基因组注释已经得到了广泛应用,但仍然存在许多技术瓶颈需要解决。在实验技术层面,蛋白质组学相对偏低的鉴定肽段覆盖度严重降低了N端的修正效率,需要采用具有针对性的技术手段对N端进行专门分析[40],而不是一味地进行大规模蛋白质组学数据产出。在信息学层面,尽管原核生物基因组中无内含子存在,采用六阅读框翻译构建数据库的方式仍然极大地提升了数据库中候选肽段数目,并且会引入相当比例的假阳性结果,致使新肽段鉴定可信度低于原有的注释肽段[50]。现在广泛使用的比较蛋白质基因组学分析策略,在搜库时加入了同源物种蛋白质序列,提高了蛋白质序列的冗余度,也会影响基于正反库的质量控制策略对错误发现率(False discovery rate, FDR) 的估计。因此在数据库构建时,应尽可能地排除完全不可能的肽段序列,在同源基因层面估计结果的FDR,并对新肽段进行单独的质控控制。在信息整合方面,从质谱数据分析到新肽段的确认与验证,一直没有合适的标准去衡量相应操作的可靠性。过多的人为操作不仅降低了结果的可重复性和可信度,也降低了分析的效率。在蛋白质组学数据通量不断提升的今天,大量微生物的蛋白质组学数据将会涌现,一套完整的高通量、自动化基因组重注释流程显得十分重要。Kumar等在对慢生型大豆根瘤菌Bradyrhizobium japonicum进行蛋白质基因组分析时,推出了半自动化分析流程Genosuite[30],可以规模化的完成基因组重注释并对结果进行展示。尽管Genosuite没有整合基因组序列预测信息与同源信息,但是其从谱图到肽段再到蛋白质的多搜索引擎整合质量控制体系也开启了蛋白质基因组学流程化分析的先河,为原核生物的批量化重注释奠定了基础。
4 小结与展望
后基因组时代,整合转录组与蛋白质组等多组学数据,明确基因的结构和功能,对于揭示相应物种的生物学特性具有十分重要的意义。蛋白质基因组学结束了蛋白质组学与基因组学独立研究的状态。利用蛋白质组学实现基因产物高通量、高肽段覆盖的鉴定,可以有效地对原有基于序列预测和同源比对的基因组注释结果进行修正。比较蛋白质基因组学策略的出现,令大量基因组测序错误得到修正,终结了基因组学独树一帜的局面。尽管现阶段蛋白质基因组学研究在质量控制与信息整合等方面还有一些问题需要解决,但在蛋白质组学数据快速产出并得以共享的大背景下[51],相应的信息学问题将很快得以解决。未来蛋白质基因组学研究将可能向3个方面迈进:一是多组学整合,即充分整合基因组、转录组与蛋白质组学的数据特征,提高基因组注释的准确性;二是规范的自动化分析流程,保证注释修正的可靠性;三是不再拘泥于原核生物,现有的质谱分离技术以及算法的不断改进,真核生物基因组注释的大规模修正也正逐步得以推广[52],并将成为未来蛋白质组学研究的热点之一。
REFERENCES
[1] Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 1995, 269(5223): 496–512.
[2] Nielsen P, Krogh A. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics, 2005, 21(24): 4322–4329.
[3] Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, et al. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res, 2005, 33(20): 6494–6506.
[4] Fickett JW. The gene identification problem: an overview for developers. Comput Chem, 1996, 20(1): 103–118.
[5] Mathe C, Sagot MF, Schiex T, et al. Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res, 2002, 30(19): 4103–4117.
[6] Poptsova MS, Gogarten JP. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology, 2010, 156(Pt 7): 1909–1917.
[7] Korf I. Genomics: the state of the art in RNA-seq analysis. Nat Methods, 2013, 10(12): 1165–1166.
[8] Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature, 2003, 422(6928): 198–207.
[9] Castellana NE, Shen Z, He Y, et al. An automated proteogenomic method utilizes mass spectrometry to reveal novel genes in zea mays. Mol Cell Proteomics, 2014, 13(1): 157–167.
[10] Christie-Oleza JA, Miotello G, Armengaud J. High-throughput proteogenomics of Ruegeria pomeroyi: seeding a better genomic annotation for the whole marine Roseobacter clade. BMC Genomics, 2012, 13: 73.
[11] Castellana N, Bafna V. Proteogenomics to discover the full coding content of genomes: a computational perspective. J Proteomics, 2010, 73(11): 2124–2135.
[12] Delcher AL, Bratke KA, Powers EC, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics, 2007, 23(6): 673–679.
[13] Lukashin AV, Borodovsky M. GeneMark. hmm: new solutions for gene finding. Nucleic Acids Res, 1998, 26(4): 1107–1115.
[14] Raghavan R, Sage A, Ochman H. Genome-wide identification of transcription start sites yields a novel thermosensing RNA and new cyclic AMP receptor protein-regulated genes in Escherichia coli.J Bacteriol, 2011, 193(11): 2871–2874.
[15] Mendoza-Vargas A, Olvera L, Olvera M, et al. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS ONE, 2009, 4(10): e7526.
[16] Mou X, Sun S, Edwards RA, et al. Bacterial carbon processing by generalist species in the coastal ocean. Nature, 2008, 451(7179): 708–711.
[17] Denoeud F, Aury JM, Da Silva C, et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol, 2008, 9(12): R175.
[18] Stanke M, Schoffmann O, Morgenstern B, et al. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 2006, 7: 62.
[19] Wang T, Cui Y, Jin J, et al. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res, 2013, 41(9): 4743–4754.
[20] Hather G, Higdon R, Bauman A, et al. Estimating false discovery rates for peptide and protein identification using randomized databases. Proteomics, 2010, 10(12): 2369–2376.
[21] Reiter L, Claassen M, Schrimpf SP, et al. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics, 2009, 8(11): 2405–2417.
[22] Wu S, Li N, Ma J, et al. First proteomic exploration of protein-encoding genes on chromosome 1 in human liver, stomach, and colon. J Proteome Res, 2013, 12(1): 67–80.
[23] Marko-Varga G, Omenn GS, Paik YK, et al. A first step toward completion of a genome-wide characterization of the human proteome. J Proteome Res, 2013, 12(1): 1–5.
[24] Chang C, Li L, Zhang C, et al. Systematic analyses of the transcriptome, translatome, and proteome provide a global view and potential strategy for the C-HPP. J Proteome Res, 2013, (In revised).
[25] Kelkar DS, Kumar D, Kumar P, et al. Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol Cell Proteomics, 2011, 10(12): M111. 011627.
[26] Ketteler R. On programmed ribosomal frameshifting: the alternative proteomes. Front Genet, 2012, 3: 242.
[27] Kyrpides NC. Fifteen years of microbial genomics: meeting the challenges and fulfilling the dream. Nat Biotechnol, 2009, 27(7): 627–632.
[28] Yandell M, Ence D. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet, 2012, 13(5): 329–342.
[29] Li N, Wu S, Zhang C, et al. PepDistiller: a quality control tool to improve the sensitivity and accuracy of peptide identifications in shotgun proteomics. Proteomics, 2012, 12(11): 1720–1725.
[30] Kumar D, Yadav AK, Kadimi PK, et al. Proteogenomic analysis of Bradyrhizobium japonicum USDA110 using genosuite, an automated multi-algorithmic pipeline. Mol Cell Proteomics, 2013, 12(11): 3388–3397.
[31] Bachman J. Reverse-transcription PCR (RT-PCR). Methods Enzymol, 2013, 530: 67–74.
[32] Afzal V, Huang JT, Atrih A, et al. PChopper: high throughput peptide prediction for MRM/SRM transition design. BMC Bioinformatics, 2011, 12: 338.
[33] Yates JR 3rd, Eng JK, McCormack AL. Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal Chem, 1995, 67(18): 3202–3210.
[34] Jaffe JD, Berg HC, Church GM. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics, 2004, 4(1): 59–77.
[35] Baudet M, Ortet P, Gaillard JC, et al. Proteomics-based refinement of Deinococcus deserti genome annotation reveals an unwonted use of non-canonical translation initiation codons. Mol Cell Proteomics, 2010, 9(2): 415–426.
[36] Gupta N, Tanner S, Jaitly N, et al. Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation. Genome Res, 2007, 17(9): 1362–1377.
[37] Chen Z, Wen B, Wang Q, et al. Quantitative proteomics reveals the temperature-dependent proteins encoded by a series of cluster genes in thermoanaerobacter tengcongensis. Mol Cell Proteomics, 2013, 12(8): 2266–2277.
[38] Savidor A, Donahoo RS, Hurtado-Gonzales O, et al.Expressed peptide tags: an additional layer of data for genome annotation. J Proteome Res, 2006, 5(11): 3048–3058.
[39] de Groot A, Dulermo R, Ortet P, et al. Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet, 2009, 5(3): e1000434.
[40] Gallien S, Perrodou E, Carapito C, et al. Ortho-proteogenomics: multiple proteomes investigation through orthology and a new MS-based protocol. Genome Res, 2009, 19(1): 128–135.
[41] Krug K, Carpy A, Behrends G, et al. Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Mol Cell Proteomics, 2013, 12(11): 3420–3430.
[42] Denef VJ, Kalnejais LH, Mueller RS, et al. Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc Natl Acad Sci USA, 2010, 107(6): 2383–2390.
[43] Gupta N, Benhamida J, Bhargava V, et al. Comparative proteogenomics: combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res, 2008, 18(7): 1133–1142.
[44] Alexova R, Haynes PA, Ferrari BC, et al. Comparative protein expression in different strains of the bloom-forming cyanobacterium Microcystis aeruginosa. Mol Cell Proteomics, 2011, 10(9): M110 003749.
[45] Schrimpe-Rutledge AC, Jones MB, Chauhan S, et al. Comparative omics-driven genome annotation refinement: application across Yersiniae. PLoS ONE, 2012, 7(3): e33903.
[46] Zhong Y, Chang X, Cao XJ, et al. Comparative proteogenomic analysis of the Leptospira interrogans virulence-attenuated strain IPAV against the pathogenic strain 56601. Cell Res, 2011, 21(8): 1210–1229.
[47] Plewniak F, Bianchetti L, Brelivet Y, et al. PipeAlign: a new toolkit for protein family analysis. Nucleic Acids Res, 2003, 31(13): 3829–3832.
[48] Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res, 2004, 32(5): 1792–1797.
[49] Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol, 2001, 314(5): 1041–1052.
[50] Blakeley P, Overton IM, Hubbard SJ. Addressing statistical biases in nucleotide-derived protein databases for proteogenomic search strategies. J Proteome Res, 2012, 11(11): 5221–5234.
[51] Vizcaino JA, Cote RG, Csordas A, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res, 2013, 41(Database issue): D1063–1069.
[52] Branca RM, Orre LM, Johansson HJ, et al. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat Methods, 2014, 11(1): 59–62.
(本文责编 陈宏宇)
Progress in proteogenomics of prokaryotes
Chengpu Zhang, Ping Xu, and Yunping Zhu
Beijing Proteome Research Center, State Key Laboratory of Proteomics, National Engineering Research Center for Protein Drugs, National Center for Protein Sciences Beijing, Beijing Institute of Radiation Medicine, Beijing 102206, China
With the rapid development of genome sequencing technologies, a large amount of prokaryote genomes have been sequenced in recent years. To further investigate the models and functions of genomes, the algorithms for genome annotations based on the sequence and homology features have been widely implemented to newly sequenced genomes. However, gene annotations only using the genomic information are prone to errors, such as the incorrect N-terminals and pseudogenes. It is even harder to provide reasonable annotating results in the case of the poor genome sequencing results. The transcriptomics based on the technologies such as microarray and RNA-seq and the proteomics based on the MS/MS have been used widely to identify the gene products with high throughput and high sensitivity, providing the powerful tools for the verification and correction of annotated genome. Compared with transcriptomics, proteomics can generate the protein list for the expressed genes in the samples or cells without any confusion of the non-coding RNA, leading the proteogenomics an important basis for the genome annotations in prokaryotes. In this paper, we first described the traditional genome annotation algorithms and pointed out the shortcomings. Then we summarized the advantages of proteomics in the genome annotations and reviewed the progress of proteogenomics in prokaryotes. Finally we discussed the challenges and strategies in the data analyses and potential solutions for the developments of proteogenomics.
proteogenomics, prokaryotes, genome annotation, mass spectrometry
December 24, 2013; Accepted: February 17, 2014
Yunping Zhu. Tel/Fax: +86-10-80705225; E-mail: zhuyunping@gmail.com
张成普, 徐平, 朱云平. 原核生物蛋白质基因组学研究进展. 生物工程学报, 2014, 30(7): 1026−1035.
Zhang CP, Xu P, Zhu YP. Progress in proteogenomics of prokaryotes. Chin J Biotech, 2014, 30(7): 1026−1035.
Supported by: National Basic Research Program of China (973 Program) (Nos. 2011CB910600, 2010CB912700, 2013CB911200), National High Technology Research and Development Program of China (863 Program) (Nos. 2012AA020409, 2012AA020201), National Natural Science Foundation of China (Nos. 21105121, 21275160), Beijing Natural Science Foundation (No. 5122013).
Ping Xu. Tel: +86-10-83147777-1314; Fax: +86-10-80705155; E-mail: xupingghy@gmail.com
国家重点基础研究计划 (973计划) (Nos. 2011CB910600,2010CB912700,2013CB911200),国家高技术研究发展计划 (863计划) (Nos. 2012AA020409, 2012AA020201),国家自然科学基金 (Nos. 21105121, 21275160),北京市自然科学基金 (No. 5122013) 资助。
时间:2014-03-25 网络出版地址:http://www.cnki.net/kcms/doi/10.13345/j.cjb.130659.html
