高精度相对和绝对定量的等量异位标签在定量蛋白质组学研究中的新进展
2014-06-15马首智孙玉琳赵晓航徐平
马首智,孙玉琳,赵晓航,徐平
高精度相对和绝对定量的等量异位标签在定量蛋白质组学研究中的新进展
马首智1,孙玉琳1,赵晓航1,徐平2,3
1 北京协和医学院 中国医学科学院肿瘤医院 分子肿瘤学国家重点实验室,北京 100021 2 军事医学科学院放射医学研究所,北京 100850 3 北京蛋白质组研究中心,北京 102206
蛋白质组学逐渐从定性研究转向定量研究。在定量蛋白质组学技术中,相对和绝对定量的等量异位标签 (Isobaric tags for relative and absolute quantitation, iTRAQ) 是应用最广泛的技术之一,具有通量高、稳定性强及不受样品来源制约等优点,几乎可以对任意样品进行标记,而且可以同时对多达8个样品进行定量分析,有效地提高了通量。iTRAQ技术不断改进,其定量准确性显著提高,适用的平台越来越多,为微生物、动物、植物、生物医学领域蛋白质及其翻译后修饰组研究创造了条件。文中综述了高精度iTRAQ技术在定量蛋白质组学研究中的最新发展及其应用。
定量蛋白质组学,相对和绝对定量的等量异位标签,定量精确度
蛋白质组学是一门新兴学科,主要目的是从整体水平上研究蛋白的组成及其生物学功能。与传统生物学研究针对单个蛋白不同,蛋白质组学同时研究数千乃至上万个蛋白在特定生物学过程中的变化。高等生物蛋白质组具有高度的动态范围和复杂性,给蛋白质组学研究带来了严峻挑战。传统的生物化学技术无法满足蛋白质组学研究的需求,需要新技术的有力支撑。蛋白质组学技术的进步可以推动蛋白质组学研究的发展,每一次重大的技术突破都会改变蛋白质组学研究的面貌。其中,iTRAQ是近年来新出现的一种蛋白质定量技术。自诞生以来,iTRAQ技术得到了不断改进,在不同领域有了广泛的应用。
1 iTRAQ技术原理
iTRAQ是美国应用生物系统公司 (Applied biosystems Inc, ABI) 的Ross等[1]于2004年发展的一种用于体外标记蛋白质或多肽的试剂。以4标试剂盒为例,该组试剂由4种相同分子量的稳定同位素标签 (Isobaric tag) 组成。每个标签包括肽反应基团 (Reactive group)、平衡基团(Balance group)和报告基团 (Reporter group) 等3部分 (图1A)。肽反应基团可与多肽的N端或赖氨酸侧链的ε氨基共价连接。报告基团的分子量分别为114、115、116、117道尔顿 (Da),平衡基团分别为31、30、29、28 Da。每个iTRAQ试剂的报告基团和平衡基团分子量加和相等,所以不同iTRAQ试剂标记的多肽在一级质谱中分子量没有差异,这种设计不仅可以增强质谱信号,还可有效地减少混合样品的复杂性。在二级质谱中,这3个基团之间发生断裂,平衡基团由于中性丢失不能被检测;报告基团可以被质谱检测到,因而可以用报告基团区分样品的来源。由于这些不同样品来源但相同质荷比的母离子可被选择进行高能碰撞裂解,释放出代表来源样品的不同质荷比的报告离子和完全一致的子离子系列。这些子离子系列包含有氨基酸序列信息,可用于蛋白质鉴定。而这些在同一个二级谱图中包含的报告离子的丰度代表了不同来源样品中该特定蛋白的多少,可用于蛋白质定量分析。
2 iTRAQ技术发展趋势
iTRAQ技术因其独特的技术优势,问世后受到了蛋白质组学界的广泛关注,并在应用中得以不断地改进和优化。iTRAQ技术包括了3个方面的发展趋势,即通量不断提高、适用的质谱平台逐渐增加以及iTRAQ的定量准确性显著改善。
2.1 iTRAQ通量的提高
Pierce等[2]通过对分子结构的改进,设计出可以同时标记8个样品的iTRAQ试剂,具体结构并未公开(图1B)。与iTRAQ原理类似的试剂还有Thermo公司的串联质谱标记(Tandem mass tags, TMT)试剂[3]。TMT试剂可以同时标记6个样品(图1C)。不同标记试剂对蛋白鉴定的影响不同。Pichler等[4]研究发现在相同条件下,4重iTRAQ试剂鉴定的蛋白最多,其次是TMT试剂,8重iTRAQ试剂鉴定的蛋白最少。这主要是因为不同的iTRAQ试剂分子量不同,8重iTRAQ试剂加到肽段上的分子量更高,产生的谱图更复杂,导致常用的搜库软件,如Mascot、Sequest等搜索后给出的匹配结果变差,质谱谱图分值低,因而在质控过滤时更容易被过滤掉。因此在相同的假阳性率条件下,8重iTRAQ试剂鉴定到的蛋白数量最少。
与传统的细胞培养稳定同位素标记(Stable isotope labeling of amino acids in culture,SILAC)技术相比,iTRAQ技术可以同时分析的样品更多。最近美国威斯康辛大学的Coon研究小组发展出一种基于中子编码(Neutron encoding,NeuCode)的多重蛋白质组定量技术,它考虑了由稳定同位素原子核结合能变异引起的细微质量差异,可根据这种细微的质量差异实现不同标签标记的区分和定量比较。将NeuCode指纹引入SILAC技术,即发展为NeuCode SILAC[5]。以赖氨酸为例,这一技术理论上可以达到39重标记。它结合了iTRAQ技术高通量和SILAC技术高精度两方面的优点,可能是未来iTRAQ技术发展的一个趋势。
2.2 iTRAQ检测平台的发展
早期iTRAQ标记的样品使用飞行时间质谱(Time of flight,TOF) 进行检测。而在离子阱质谱(Ion trap, IT)中应用较晚,这是因为传统的离子阱质谱使用碰撞诱导解离 (Collsion induced dissociation, CID) 技术,由于四分之一效应的存在,同时也由于低质荷比端噪音高,无法检测低质荷比区域的iTRAQ报告离子。Griffin等[6]引入了脉冲Q点裂解 (Pulsed Q dissociation, PQD) 技术,使IT质谱可用于iTRAQ报告离子的检测。更高能量碰撞解离 (Higher-energy collisional dissociation, HCD) 技术[7]的引入扩展了IT质谱的检测范围,并且提高了质谱分析精度。
2.3 iTRAQ定量准确性的改善
iTRAQ技术发展的另一个趋势是不断提高蛋白定量的准确性。蛋白定量时目标蛋白产生的报告离子容易受到杂质离子产生的报告离子的干扰,导致定量准确性降低。最近2篇发表在Nature Methods上的工作采用新的质谱方法进行iTRAQ分析,试图解决这个问题。其中,Wenger等[8]将一级质谱的目标离子进行电子转移解离(Electron transfer dissociation,ETD)处理,ETD处理改善了定量效果,但并没有完全去除干扰离子对定量的影响。在另一项研究中,Ting等[9]将蛋白用LysC酶解后进行标记,二级质谱采用CID裂解但并不进行定量分析,而是选择信号最强的带有报告离子的碎片离子再进行第三级HCD裂解,通过三级质谱产生的报告离子进行定量。因为杂质产生的碎片离子与样品产生的碎片离子质荷比不同,所以很少有干扰离子能够进入三级质谱,从而使目标离子的定量准确度得到提高。使用已知比值(10∶1)的标准蛋白进行质谱分析,当没有杂质离子干扰时,蛋白的中位比值为11.7∶1,而有杂质离子干扰时,蛋白中位比值变为3.7∶1。如果采用三级质谱方法,蛋白的中位比值可恢复为10.5∶1。这说明三级质谱方法几乎彻底去除了干扰离子对定量的影响,有效地提高了定量蛋白质组的精度。
三级质谱技术出现后,又得到了不断的优化。Dayon等[10]通过将三级质谱与气相分离结合,并换用胰酶进行酶解,进一步提高了蛋白鉴定数量。三级质谱方法的质谱周期较长。对此,Wühr等[11]发展了互补TMT(Complement TMT, TMTc)的方法,根据二级质谱中TMT试剂部分断裂的产物进行定量,这些离子含有肽段和未充分裂解的TMT试剂,残余的TMT试剂含有稳定同位素,可以区分样品来源并提供定量信息。这种方法可以在不增加质谱分析时间的情况下准确定量,但并非所有的二级质谱图都含有合适的TMTc离子。
2.4 mTRAQ技术的引入
相对和绝对定量的不等量标签(Mass differential tags for relative and absolute quantification,mTRAQ)技术是与iTRAQ原理类似的一种技术。与iTRAQ的差别在于mTRAQ技术的标签并不等重(图1D、E、F)。mTRAQ设计的最初目的是用于质谱多反应监测(Multiple reaction monitoring, MRM)分析,研究发现mTRAQ也适合鸟枪法测序[12]。随后对质谱、生物信息等技术的改进提升了mTRAQ分析的质量[13-14],扩展了mTRAQ技术在定量蛋白质组学中的应用。使用2-甲氧基-4,5-二氢-1氢-咪唑也可以对酶解后的肽段进行不等量标记[15]。
3 iTRAQ技术与其他技术的比较
蛋白质组学定量技术包括基于凝胶的定量技术和基于质谱的定量技术。近年来,基于质谱的定量技术逐渐成为主流。基于质谱的定量技术可分为3类:代谢标记技术 (Metabolic labeling)、化学标记技术 (Chemical labeling)、无标记定量技术 (Label free)。3种方法的主要区别是对同位素标签的使用不同。以SILAC为代表的代谢标记技术在细胞培养阶段加入同位素。以iTRAQ技术为代表的化学标记技术对抽提的蛋白和多肽进行同位素标记。无标记定量技术则不使用同位素标签。不同标记技术各有其优缺点 (表1)。
SILAC技术要求细胞在同位素标记培养基中生长一段时间,不能在体外分裂的细胞无法进行SILAC标记。iTRAQ技术可以对肽段进行体外标记,因此对样品来源没有限制。此外,不同iTRAQ标记的肽段在一级质谱时形成一个峰,有利于蛋白鉴定。Pütz等[16]比较了iTRAQ和SILAC技术,结果表明iTRAQ鉴定到的蛋白和差异蛋白数量更多。与SILAC相比,iTRAQ的缺点是蛋白经过酶解后再进行标记,这个过程使得从蛋白质提取到消化为肽段的样品处理过程无法监控,可能引入人为干扰,增加了实验误差,影响定量比较的精度。而传统iTRAQ技术准确度较差,更加剧了这种定量蛋白质组学研究的难度。
无标记定量技术对质谱稳定性有严格要求,稳定性差的质谱平台难以得到有统计学意义的定量信息。使用iTRAQ技术可以降低对质谱的要求,而且提高了通量。例如,Wang等[17]比较了iTRAQ技术与非标记定量技术,iTRAQ的变异系数(Coefficient of variation, CV)只有1.8%,而无标记定量的CV值达到了15.6%,但在测量变化倍数高的蛋白时,传统的iTRAQ的准确性低于无标记定量技术。Li等[18]系统比较了代谢标记、化学标记(iTRAQ/TMT)和无标记定量技术。结果表明,代谢标记和化学标记的定量准确度、精确度和可重复性相当,而就定量精确性和可重复性来说,iTRAQ/TMT技术还优于代谢标记技术。但尽管如此报道,由于代谢标记的理论定量精度更高,因此需要更为细致、精确的评价。由于不同的技术方法各有其优缺点,可根据不同的实验目的选择不同的定量方法。
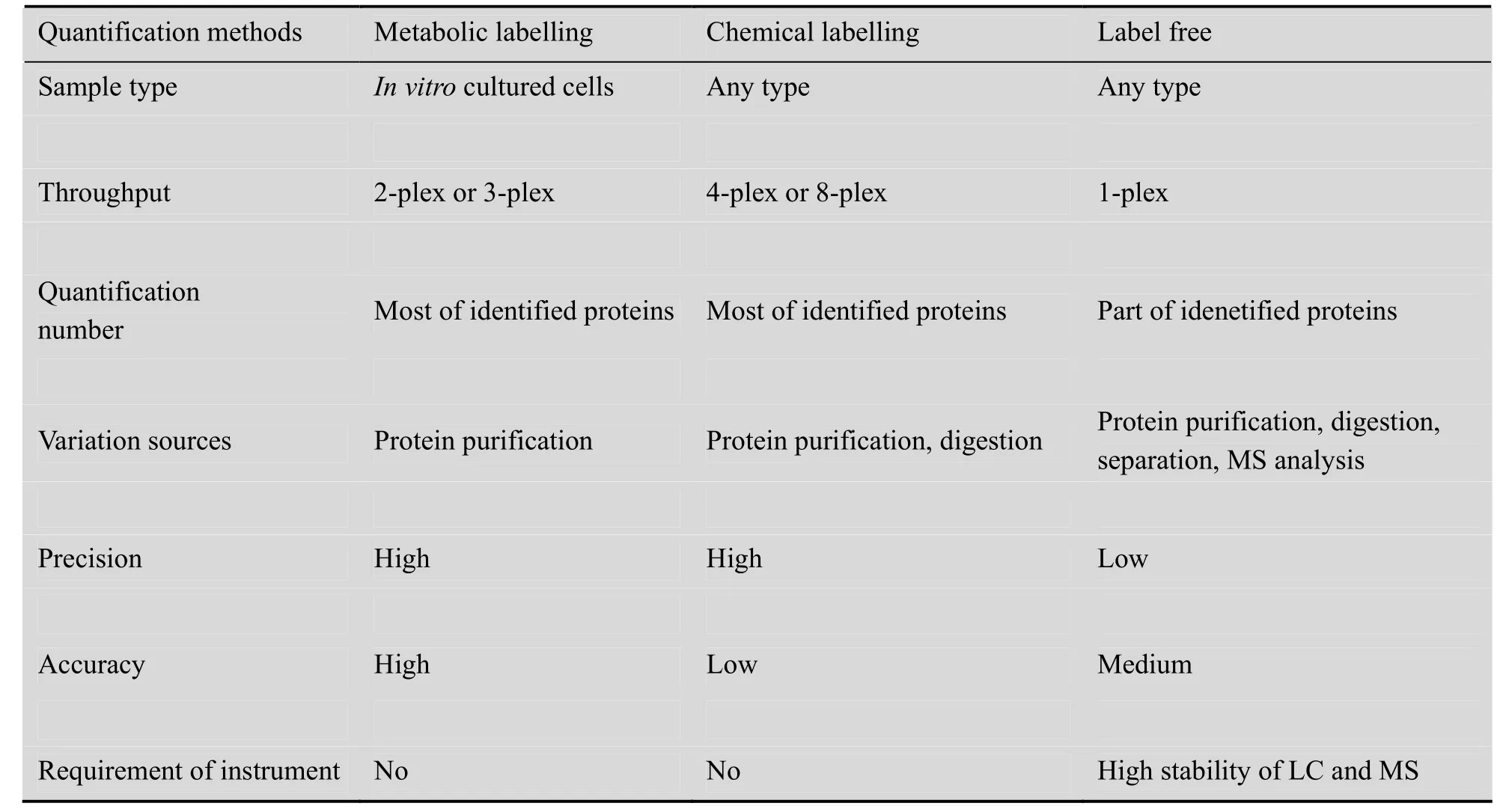
表1 不同蛋白定量方法比较Table 1 Comparasion of different quantification methods
4 iTRAQ技术的应用
随着技术的进步,iTRAQ得到了越来越广泛的应用。iTRAQ技术用于蛋白定量分析,在微生物、动物、植物、生物医学等生物材料中都有应用;此外,iTRAQ还可以用于蛋白翻译后修饰研究。
4.1 iTRAQ技术用于微生物材料
微生物结构简单,蛋白成分复杂度低,是蛋白质组学研究的理想工具。大肠杆菌是最早应用iTRAQ技术的模式生物之一[19]。此后,iTRAQ在微生物研究中的应用不断深入。Wang等[17]分析了高产油的衣藻和亲本衣藻蛋白的差别,发现在高产油的衣藻中,参与脂肪酸合成的蛋白上调,而参与淀粉合成的蛋白下调,从而促进了油脂的生成。Rivera等[20]分析了转录因子CodY对葡萄球菌毒力的影响。iTRAQ分析表明CodY敲除后葡萄球菌的蛋白酶和溶血素表达升高,增加了细菌的毒力。Lee等[21]分析了胆汁盐处理前后约氏乳杆菌蛋白质组变化,发现差异蛋白涉及胁迫响应、细胞分裂、碳水化合物代谢等,这有助于益生菌改造。此外,Komatsu等[22]将mTRAQ技术用于微生物研究,发现了抗菌蛋白histatin 5调控白色念珠菌线粒体蛋白质组的信号网络。此外,宏蛋白质组指微生物群落所产生的全部蛋白质[23],iTRAQ还可以用于宏蛋白质组学分析。
4.2 iTRAQ技术用于动物材料
动物样本难以直接进行代谢标记。在模式动物的研究中,iTRAQ可以有效解决样品标记的难题。有机汞对环境造成了严重污染,Cuello等[24]用iTRAQ技术分析了甲基汞处理前后的斑马鱼蛋白质组变化,发现71个表达改变的蛋白,分析结果表明甲基汞影响了胚胎发育、蛋白合成、能量代谢、钙离子稳态等。Sun等[25]用iTRAQ技术分析了高血压疾病模型小鼠和正常小鼠的差异蛋白,发现ATP6-VOA1、ATP6V1D、DLD、MAO-A等4个蛋白可以有效区分疾病小鼠和正常对照。
4.3 iTRAQ技术用于植物材料
植物生长过程中经常遭受恶劣环境,抗逆反应可以帮助植物对抗恶劣环境,对抗逆反应的研究有助于提高作物产量。Abdalla等[26]分析了抗旱植物维柯萨对脱水胁迫的响应,作者用模拟脱水的药物处理植物,提取了处理前后的细胞核,使用iTRAQ技术进行定量分析,鉴定到的差异蛋白涉及信号转导、核浆转运、分子伴侣等。Alvarez等[27]分析了脱落酸(Abscisic acid, ABA)处理的拟南芥GPA1突变体蛋白表达变化,结果显示ABA信号和内质网转运有重要的联系,而GPA1既在ABA信号网络中发挥作用,也有独立于ABA信号的其他功能。
4.4 iTRAQ技术用于生物医学材料
人体组织或体液是生物标记物的主要来源。人体组织样本或体液样本无法在体外分裂或扩增,而且来源有限,极为珍贵,因此iTRAQ是一种理想的标记技术,在组织样本的研究中得到了广泛应用。
Zeng等[28]检测了肺癌、转移癌、不典型增生和正常组织的蛋白表达,鉴定到102个显著变化的蛋白。后期的研究选取GSTP1、HSPB1和 CKB三个蛋白进行验证,结果表明这3个蛋白的组合对不同病变区分的灵敏度和特异度均超过90%。Mangé等[29]从接受透析治疗的患者血浆中分离高密度脂蛋白的复合体,使用iTRAQ技术鉴定相互作用蛋白,鉴定到apoC-Ⅱ、apoC-Ⅲ、serotransferrin和haptoglobin等几个蛋白,这些蛋白可以预测患者接受透析治疗时出现并发症的风险。
Kang等[30]采用mTRAQ技术从结肠癌和癌旁组织中鉴定到超过3 500个蛋白。此外,mTRAQ技术还可用于血清、福尔马林固定样品中蛋白的鉴定[31-32]。mTRAQ与MRM技术相结合,也常用于肿瘤标志物的靶向研究[33-34]。
4.5 iTRAQ技术用于蛋白质翻译后修饰
翻译后修饰对蛋白功能有重要的调控作用,iTRAQ结合特定的富集技术,可用于翻译后修饰研究。Reiter等[35]纯化了氧化应激条件下酵母的Pan1蛋白,酶解后用iTRAQ试剂进行标记,再富集磷酸化修饰多肽,结果表明Pan1的磷酸化修饰受到氧化应激的调控。Tian等[36]用固相萃取的方法纯化血清糖基化修饰蛋白,并比较了侵袭性和非侵袭性前列腺癌患者血清糖基化修饰蛋白。结果表明Cathepsin L、Periostin和MFAP4等蛋白在侵袭性肿瘤里表达升高,这些蛋白可以作为诊断侵袭性前列腺癌的标志物。Melo-Braga等[37]用不同的亲和介质富集各种翻译后修饰蛋白,结合iTRAQ标记和质谱检测,同时分析了蛋白表达谱、磷酸化修饰、糖基化修饰和乙酰化修饰的变化。他们从小卷蛾侵染的葡萄果皮中鉴定到3 059个蛋白,其中899个蛋白、110个磷酸化修饰位点、10个糖基化修饰位点和20个乙酰化修饰位点在侵染前后有显著变化。
5 展望
随着后基因组时代的到来,对基因功能的诠释成为生命科学领域研究的主要内容。定量蛋白质组学在这个过程中发挥了重要作用,定量蛋白质组学通过研究与某个功能有关的一群蛋白表达差异,建立细胞内外信号转导网络,揭示生命奥秘。iTRAQ技术以其诸多优点,在定量蛋白质组学研究中得到广泛应用。本实验室正在进行的细胞死亡调控机理研究,期望采用iTRAQ技术分析调控细胞坏死的信号网络,并从中发现新的药物靶标和化疗敏感性标志物。而这一技术本身的不断进步使其能更有效地分析生物样本,回答各种生物学问题。将来iTRAQ技术会更广泛地应用到生物学研究中,并极大地推动生物学研究的发展。
REFERENCES
[1] Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in S accharomyces c erevisiae using mine-reactive isobaric tagging reagents. Mol Cell Proteomics, 2004, 3(12): 1154–1169.
[2] Pierce A, Unwin RD, Evans CA, et al. Eight-channel iTRAQ enables comparison of the activity of six leukemogenic tyrosine kinases. Mol Cell Proteomics, 2008, 7(5): 853–863.
[3] Thompson A, Schäfer J, Kuhn K, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem, 2003, 75(8): 1895–1904.
[4] Pichler P, Köcher T, Holzmann J, et al. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal Chem, 2010, 82(15): 6549–6558.
[5] Hebert AS, Merrill AE, Bailey DJ, et al. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat Methods, 2013, 10(4): 332–334.
[6] Griffin TJ, Xie H, Bandhakavi S, et al. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J Proteome Res, 2007, 6(11): 4200–4209.
[7] Olsen JV, Macek B, Lange O, et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods, 2007, 4(9): 709–712.
[8] Wenger CD, Lee MV, Hebert AS, et al. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods, 2011, 8(11): 933–935.
[9] Ting L, Rad R, Gygi SP, et al. MS3 eliminates ratiodistortion in isobaric multiplexed quantitative proteomics. Nat Methods, 2011, 8(11): 937–940.
[10] Dayon L, Sonderegger B, Kussmann M. Combination of gas-phase fractionation and MS(3) acquisition modes for relative protein quantification with isobaric tagging. J Proteome Res, 2012, 11(10): 5081–5089.
[11] Wühr M, Haas W, McAlister GC, et al. Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal Chem, 2012, 84(21): 9214–9221.
[12] Kang UB, Yeom J, Kim H, et al. Quantitative analysis of mTRAQ-labeled proteome using full MS scans. J Proteome Res, 2010, 9(7): 3750–3758. [13] Yoon JY, Lim KY, Lee S, et al. Improved quantitative analysis of mass spectrometry using quadratic equations. J Proteome Res, 2010, 9(5): 2775–2785.
[14] Yoon JY, Yeom J, Lee H, et al. High-throughput peptide quantification using mTRAQ reagent triplex. BMC Bioinformatics, 2011, 12(Suppl 1): S46.
[15] Gao DM, Sun L, Guo K, et al. Evaluation of isotopic labeling of lysine residues of peptides for quantitative proteomics. Chin J Biotech, 2012, 28(7): 855–864 (in Chinese).高东梅, 孙璐, 郭坤, 等. 评价新型稳定同位素赖氨酸标记在定量蛋白质组学中的应用. 生物工程学报, 2012, 28(7): 855−864.
[16] Pütz SM, Boehm AM, Stiewe T, et al. iTRAQ analysis of a cell culture model for malignant transformation, including comparison with 2D-PAGE and SILAC. J Proteome Res, 2012, 11(4): 2140–2153.
[17] Wang H, Alvarez S, Hicks LM. Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J Proteome Res, 2012, 11(1): 487–501.
[18] Li Z, Adams RM, Chourey K, et al. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res, 2012, 11(3): 1582–1590.
[19] Choe LH, Aggarwal K, Franck Z, et al. A comparison of the consistency of proteome quantitation using two-dimensional electrophoresis and shotgun isobaric tagging in Escherichia coli cells. Electrophoresis, 2005, 26(12): 2437–2449.
[20] Rivera FE, Miller HK, Kolar SL, et al. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics, 2012, 12(2): 263–268.
[21] Lee JY, Pajarillo EA, Kim MJ, et al. Proteomic and transcriptional analysis of Lactobacillus j ohnsonii PF01 during bile salt exposure by iTRAQ shotgun proteomics and quantitative RT-PCR. J Proteome Res, 2013, 12(1): 432–443.
[22] Komatsu T, Salih E, Helmerhorst EJ, et al. Influence of histatin 5 on Candida a lbicans mitochondrial protein expression assessed by quantitative mass spectrometry. J Proteome Res, 2011, 10(2): 646–655.
[23] Yu RT, Gao PJ, Han L, et al. Strategy and application of metaproteomics. Chin J Biotech, 2009, 25(7): 961–967 (in Chinese).于仁涛, 高培基, 韩黎, 等. 宏蛋白质组学研究策略及应用. 生物工程学报, 2009, 25(7): 961−967.
[24] Cuello S, Ximénez-Embún P, Ruppen I, et al. Analysis of protein expression in developmental toxicity induced by MeHg in zebrafish. Analyst, 2012, 137(22): 5302–5311.
[25] Sun D, Cheng Y, Zhou D, et al. Quantitative proteome of medulla oblongata in spontaneously hypertensive rats. J Proteome Res, 2013, 12(1): 390–395.
[26] Abdalla KO, Rafudeen MS. Analysis of the nuclear proteome of the resurrection plant Xerophyta viscosa in response to dehydration stress using iTRAQ with 2DLC and tandem mass spectrometry. J Proteomics, 2012, 75(8): 2361–2374.
[27] Alvarez S, Hicks LM, Pandey S. ABA-dependentand -independent G-protein signaling in Arabidopsis roots revealed through an iTRAQ proteomics approach. J Proteome Res, 2011, 10(7): 3107–3122.
[28] Zeng GQ, Zhang PF, Deng X, et al. Identification of candidate biomarkers for early detection of human lung squamous cell cancer by quantitative proteomics. Mol Cell Proteomics, 2012, 11(6): M111.013946.
[29] Mangé A, Goux A, Badiou S, et al. HDL proteome in hemodialysis patients: a quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE, 2012, 7(3): e34107.
[30] Kang UB, Yeom J, Kim HJ, et al. Expression profiling of more than 3500 proteins of MSS-type colorectal cancer by stable isotope labeling and mass spectrometry. J Proteomics, 2012, 75(10): 3050–3062.
[31] Suh EJ, Kabir MH, Kang UB, et al. Comparative profiling of plasma proteome from breast cancer patients reveals thrombospondin-1 and BRWD3 as serological biomarkers. Exp Mol Med, 2012, 44(1): 36–44.
[32] DeSouza LV, Krakovska O, Darfler MM, et al. mTRAQ-based quantification of potential endometrial carcinoma biomarkers from archived formalin-fixed paraffin-embedded tissues. Proteomics, 2010, 10(17): 3108–3116.
[33] DeSouza LV, Taylor AM, Li W, et al. Multiple reaction monitoring of mTRAQ-labeled peptides enables absolute quantification of endogenous levels of a potential cancer marker in cancerous and normal endometrial tissues. J Proteome Res, 2008, 7(8): 3525–3534.
[34] DeSouza LV, Romaschin AD, Colgan TJ, et al. Absolute quantification of potential cancer markers in clinical tissue homogenates using multiple reaction monitoring on a hybrid triple quadrupole/linear ion trap tandem mass spectrometer. Anal Chem, 2009, 81(9): 3462–3470.
[35] Reiter W, Anrather D, Dohnal I, et al. Validation of regulated protein phosphorylation events in yeast by quantitative mass spectrometry analysis of purified proteins. Proteomics, 2012, 12(19/20): 3030–3043.
[36] Tian Y, Bova GS, Zhang H. Quantitative glycoproteomic analysis of optimal cutting temperature-embedded frozen tissues identifying glycoproteins associated with aggressive prostate cancer. Anal Chem, 2011, 83(18): 7013–7019.
[37] Melo-Braga MN, Verano-Braga T, León IR, et al. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol Cell Proteomics, 2012, 11(10): 945–956.
(本文责编 陈宏宇)
马首智, 孙玉琳, 赵晓航, 等. 高精度相对和绝对定量的等量异位标签在定量蛋白质组学研究中的新进展. 生物工程学报, 2014, 30(7): 1073−1082.
Ma SZ, Sun YL, Zhao XH, et al. Recent advance in high accuracy iTRAQ for quantitative proteomics. Chin J Biotech, 2014, 30(7): 1073−1082.
Recent advance in high accuracy iTRAQ for quantitative proteomics
Shouzhi Ma1, Yulin Sun1, Xiaohang Zhao1, and Ping Xu2,3
1 State Key Laboratory of Molecular O ncology, Cancer Hospital & Institute, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing 100021, China 2 Institute of Radiation Medicine, Academy of Military Medical Sciences, Beijing 100850, China 3 Beijing Proteome Research Center, Beijing 102206, China
Nowadays, proteomics focuses on quantitative analysis rather than qualitative. In the field of quantitative proteomics, Isobaric tags for relative and absolute quantitation (iTRAQ) is one of the most widely used techniques. The advantage of iTRAQ is high throughput, high stability and free of the restriction of sample property. iTRAQ is suitable for almost all kinds of samples, and up to 8 samples can be analyzed simultaneously by commercially available kit. Along with the development of techniques, more and more mass spectrometry (MS) platforms are used in iTRAQ experiments and the accuracy of iTRAQ has been improved. iTRAQ has been applied to studies of microorganism, animal, plant, medical and protein post-translational modification. Here we review the recent progress in the development of iTRAQ and its applications in quantitative proteomics.
quantitative proteomics, isobaric tags for relative and absolute quantitation, accuracy
December 18, 2013; Accepted: January 27, 2014
Ping Xu. Tel: +86-10-83147777-1314; E-mail: xupingghy@gmail.com Xiaohang Zhao. Tel: +86-10-67709015; Fax: +86-10-87778360; E-mail: zhaoxh@cicams.ac.cn
Supported by: National High Technology Research and Development Program of China (863 Program) (No. 2012AA020206), National Natural Science Foundation of China (Nos. 31070673, 31170780, 91029725, 81071789), Beijing Natural Science Foundation (No. 5112012).
国家高技术研究发展计划 (863计划) (No. 2012AA020206),国家自然科学基金 (Nos. 31070673, 31170780, 91029725, 81071789),
北京市自然科学基金 (No. 5112012) 资助。
