丙烷无氧脱氢制丙烯工艺和催化剂的研究进展
2014-05-14董秀芹余英哲张敏华
刘 乔,董秀芹,余英哲,张敏华
(1. 天津大学 绿色合成与转化教育部重点实验室,天津 300072;2. 天津大学 石油化工技术开发中心,天津 300072)
丙烯作为一种重要的石油化工原料,广泛用于生产聚丙烯、丙烯醛、丙烯酸、甘油、异丙醇、聚丙烯腈和丁辛醇等石化产品,其中用于聚丙烯生产的比例高达65%。随着市场经济的发展,这些下游衍生物的需求量迅速增长,因此也带动了丙烯的巨大需求[1-5]。
目前,全球约56%的丙烯来自蒸汽裂解装置,33%来自炼油厂催化裂化装置,剩余的则来自丙烷脱氢技术及其他反应[6]。丙烯的生产能力远远低于其需求量,且蒸汽裂解法受丙烷-乙烯联产比例的限制、催化裂化法受轻质烃进一步制取高辛烷值汽油的制约[7],因此提高丙烯生产技术已成为石化行业的主要发展趋势。扩大丙烯来源的生产工艺主要有丙烷脱氢制丙烯、甲醇制烯烃和烯烃复分解等工艺,其中丙烷脱氢制丙烯工艺的研究备受关注,已成为当代石油化工领域的一个重大课题。
本文概述了目前已工业化的丙烷无氧脱氢制丙烯技术,对常用的Cr系和Pt系催化剂进行了介绍,重点介绍了Pt系催化剂上丙烷无氧脱氢制丙烯的反应机理以及Pt系催化剂的结构、制备方法、失活原因及其应用前景。
1 丙烷无氧脱氢制丙烯工艺及其催化剂
1.1 丙烷无氧脱氢工艺
目前世界上已工业化的5种丙烷无氧脱氢制丙烯的工艺见表1。

表1 已工业化的丙烷无氧脱氢工艺Table 1 Industrialized technologies for the anaerobic dehydrogenation of propane
从表1可看出,这5种工艺在生产条件、催化剂、反应器、丙烯选择性等方面均有所不同。其中,UOP公司的Olef l ex连续移动床工艺使用最多,该工艺采用Pt系催化剂,脱氢工艺分为反应、产品回收和催化剂再生3个部分,以H2作为原料的稀释剂,在4台移动床径流式反应器中丙烷连续反应转化为丙烯,脱氢反应的转化率和选择性皆得以优化。该工艺的特点是脱氢反应均匀稳定、催化剂可在等温条件下再生等。目前,在天津渤化集团石化公司建立了全球最大的丙烷无氧脱氢装置,采用Lummus公司的Catofin工艺,该工艺使用Cr2O3/Al2O3催化剂,可生产多种烯烃(如丙烯、正丁烯、异丁烯和丁二烯)。该工艺分为4道工序(脱氢、反应器排放料压缩、产品回收和精制),采用多个固定床反应器,按循环方式操作使主物料实现连续流动。1996年,该工艺开始应用逆流式技术改变了反应物料的流向,空气向下流动,烃类物料向上流动,从而可用较少的原料获得较多的产品。STAR工艺采用PtSn/ZnAl2O4催化剂,通过两步脱氢将丙烷转化为丙烯。该反应所需热量通常从反应器外面传导引入,并利用蒸汽稀释反应气,蒸汽的存在降低了反应物和产物的分压,提高了平衡转化率,还可抑制催化剂结焦,延长催化剂的使用寿命。Linde工艺采用Cr2O3/Al2O3催化剂和固定床管式反应器,原料不需H2或蒸汽稀释,且动力消耗较低、投资较少。FBD工艺采用催化剂可连续再生的等温流化床反应器,在分离再生区被加热的催化剂为反应段中的脱氢反应提供热量。由于在流化床内使用含Cr的催化剂,所以必须注意在产物和再生器烟道气里分离催化剂粉尘的问题。
除上述常见的几种脱氢工艺外,还有尚未实现工业化的无机膜催化脱氢工艺[8]。该工艺利用不同物种在反应器内扩散速率不同的特点克服动力学平衡限制,同时催化剂还可保持高的活性和选择性,在较宽的温度范围内,使用多孔膜反应器的丙烯收率高于固定床反应器,且大于丙烯的平衡收率。该工艺虽然尚未实现工业化,但其应用前景可观,目前已引起研究者的极大兴趣。
1.2 丙烷无氧脱氢反应及其催化剂
丙烷无氧脱氢的主反应见式(1),副反应见式(2)~(7)[9];主反应的热力学数据见表2[10]。

由式(1)和表2可知,丙烷无氧脱氢主反应是强吸热且分子数增大的反应,从热力学角度分析,若使该反应向脱氢方向移动,须升高温度和降低压力。但当温度过高时,丙烷裂解、丙烯深度脱氢等副反应加剧,副产物甲烷、乙烯、乙烷的生成导致丙烯的选择性降低;且高温将加速碳在催化剂表面沉积而导致催化剂迅速失活。因此,针对丙烷无氧脱氢反应的特点,从催化剂设计和开发角度出发,该反应的关键在于开发活性、选择性及稳定性高的催化剂。
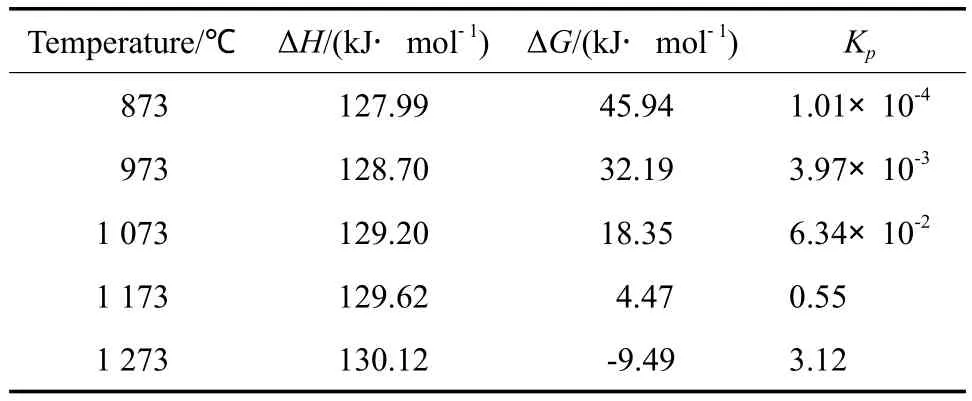
表2 丙烷无氧脱氢反应的热力学性质Table 2 Thermodynamic data for the anaerobic dehydrogenation of propane
目前工业上丙烷无氧脱氢催化剂主要为Pt系和Cr系催化剂,Catofin,Linde,FBD工艺使用的都是Cr系催化剂,且其一般是负载在稳定性较高的Al2O3上,使用较多的是具有高活性和高选择性的Cr2O3-K2O/Al2O3催化剂。对Cr系催化剂的研究主要集中在丙烷脱氢的活性位点上,Cr5+化合物的数量与催化剂的初始活性相关,但主活性中心是Cr2+,而丙烯的选择性主要由Cr3+的活性位决定[11]。但高温下催化剂的活性位易被丙烷或反应中间体裂解生成的碳覆盖,生成积碳,导致催化剂迅速失活,因此要在短周期内(15 min左右)对催化剂进行氧化除碳,因此增加了操作成本。此外,Cr系催化剂在高温下不稳定,且高价Cr对人体健康和环境保护不利,导致该类催化剂的使用受到限制。
Pt系催化剂的优势体现在高活性、高选择性、热稳定性好、低污染、低磨损率且可在苛刻条件下操作[12],但使用传统制备方法制备负载型催化剂很难控制金属粒子的分散度、粒径分布和表面形貌等微观结构,因而催化剂性能不稳定。由于丙烷催化脱氢是强吸热、分子体积增大的反应,通常需在高温低压下进行,高温使催化剂快速失活而缩短了催化剂的再生周期。为提高Pt系催化剂的选择性和稳定性,需对催化剂进行改性从而提高其抗积碳和烧结的能力。目前对Pt系催化剂改性的主要方法有加入助剂、改变催化剂载体和催化剂的制备方法等。
2 Pt系催化剂
2.1 丙烷脱氢反应机理
少量的贵金属(如Pt,Ru,Re等)分散于具有高比表面积的Al2O3和SiO2等载体上具有较高的催化活性[13]。在Pt系催化体系中,单个Pt金属原子为丙烷无氧脱氢反应的活性中心,脱氢反应的控制步骤是β-H的解离[14]。首先丙烷中的C—H 键断裂,形成吸附态的丙基;然后吸附态丙基中的一个C—H键断裂,产生一个π键,形成吸附态的丙烯;最后吸附态的丙烯脱附形成丙烯。Pt系催化剂上丙烷无氧脱氢反应机理见图1[14]。

图1 Pt系催化剂上丙烷无氧脱氢反应机理Fig.1 Reaction mechanism for the anaerobic dehydrogenation of propane on a Pt catalyst.□ Active site
2.2 催化剂粒径的影响
丙烷无氧脱氢反应是非均相催化反应,催化剂上具有活性的是其表面上的Pt原子。催化剂表面Pt原子的比例和催化剂粒径密切相关。一般用分散度(D)定义催化剂表面Pt原子在催化剂中所有原子中所占比例。如式(8)所示:

式中,NS表示催化剂表面Pt原子数目;NT表示催化剂中总的原子数目。Somorjai等[15]认为,对于小的Pt团簇(原子数为3~4),其表面Pt原子的比例高达100%。不同Pt原子数的立方团簇的结构见图2。

图2 Pt原子数为8, 27, 64, 125, 216的立方团簇结构Fig.2 Clusters of Pt atoms with single cubic packing having 8,27,64,125 and 216 atoms.
由图2可看出,粒径小的纳米粒子的分散度为1,这表示它在催化反应过程中Pt原子的利用率为100%;随Pt原子数的增加,部分Pt原子会被周围其他原子包围,成为体相原子,分散度逐渐减小。在丙烷无氧脱氢反应过程中,催化剂分散度的提高有利于提高丙烯的选择性。因此,在制备Pt系催化剂时,为提高其利用率,催化剂的粒径一般要控制在10 nm以内。
Kumar等[16]认为,对于丙烷无氧脱氢催化体系,Pt系催化剂的粒径对脱氢反应过程至关重要。主要表现在:1)脱氢反应对催化剂粒径十分敏感;2)催化剂粒径对丙烯的选择性影响非常大。在小颗粒催化剂上,丙烷无氧脱氢反应活性非常高,但催化剂上也很易生成积碳,致使丙烯选择性降低;对于大颗粒催化剂,虽丙烷转化率较低,但丙烯选择性却很高,且催化剂表面结焦量非常少。这是因为:小颗粒催化剂对C—C键断裂反应的选择性较好;大颗粒催化剂对C—H键断裂反应的选择性较好。丙烯的选择性取决于这两种断键反应的活性。
2.3 助剂
Pt系催化剂用于丙烷无氧脱氢反应的主要缺陷是稳定性差、选择性低,通过添加助剂进行修饰可明显改善其脱氢性能。其中,Sn助剂具有独特的作用,加入Sn助剂可较大幅度地优化Pt系催化剂的脱氢性能,因此引起了研究者的广泛关注。
Kumar等[17]用微湿含浸法制备了Pt/SBA-15和PtSn/SBA-15催化剂,并通过H2吸附、N2吸附、XRD和TEM-EDXS等手段进行表征,考察了Sn助剂的加入对丙烷脱氢反应的影响。他们认为,加入Sn后形成了Pt-Sn合金,提高了Pt颗粒的分散度。虽然PtSn/SBA-15催化剂表现出更高的丙烷转化率、丙烯选择性和稳定性,但Sn助剂的引入也使得催化剂结焦量明显增加,达到未加入Sn之前的3倍。Zeeshan等[18]研究了Sn助剂对PtSn/SAPO-34催化剂丙烷选择性脱氢制丙烯反应性能的影响。实验结果表明,添加适量的Sn(w=1%)在很大程度上改善了催化剂的催化性能,Sn助剂改变了金属和载体之间的相互作用,使得Pt具有良好的分散度,且Sn的加入促使生成的积碳从金属活性位转移到载体上;但Sn添加量超过1%(w)后,反而对催化性能不利。SAPO-34载体负载金属催化剂的H2-TPR表征结果见图3[18]。由图3可看出,SAPO-34载体没有出现还原峰,而Pt/SAPO-34催化剂在450~600℃内出现了尖锐的还原峰,这表明Pt与SAPO-34载体之间有强的相互作用。当加入Sn后,PtSn/SAPO-34催化剂在360~540 ℃内出现了较宽的还原峰,这表明Pt和Sn负载于SAPO-34载体上,二者的相互作用有利于脱氢反应的进行。他们还发现,Sn0能与Pt组分形成Pt-Sn合金,使Pt表面中毒而导致不可逆失活;而Sn2+可提高Pt晶粒的分散度,降低氢解活性,因此氧化态的Sn2+对Pt的催化性能起促进作用。Zhang等[19]制备了不同Sn含量的PtSn/ZSM-5催化剂并将之用于丙烷脱氢体系。研究结果表明,助剂Sn的加入不仅具有“几何效应”,减少了催化剂表面Pt颗粒的团聚,而且还改变了金属Pt与载体之间的作用。

图3 SAPO-34载体负载金属催化剂的H2-TPR表征结果Fig.3 H2-TPR curves of the SAPO-34 supported catalysts.
余长林等[20]研究了在Pt/γ-Al2O3催化剂中添加一些金属氧化物(如Ce,Sn,Zn,V,La,Cr,Fe,Zr,Mn的氧化物)对丙烷脱氢反应的影响。研究结果表明,加入CeO2,SnO2,ZrO2这3种助剂后,Pt/γ-Al2O3催化剂的脱氢活性明显提高,丙烷的初始转化率分别提高了9.2%,8.2%,8.1%。此外,Pt/γ-Al2O3催化剂的稳定性可通过添加SnO2来实现。
为进一步提高PtSn负载型催化剂的稳定性和选择性,许多学者在此基础上展开了对第二种甚至第三种助剂的研究。Huang等[21]用连续浸渍法制备了不同Sr含量的PtSnSr/HZSM-5催化剂,并采用BET,TEM,NH3-TPD,TG等手段进行了表征。表征结果显示,Sr的加入提高了Pt的分散度,适量Sr(w=1.2%)的加入提高了催化剂的活性并可有效减少催化剂的结焦量;此外他们还发现,添加第三种助剂Na(w=1.0%)时,PtSnNaSr/HZSM-5催化剂表现出优异的活性,反应5 h后,丙烯的选择性仍大于95%,丙烷的转化率为32.2%。张一卫等[22]研究了稀土金属助剂La改性对 PtSnNa/ZSM-5催化剂脱氢性能的影响,少量的La中和了催化剂表面的部分弱酸中心和强酸中心,改善了催化剂的选择性和稳定性;但过量的La会减少催化剂表面的Pt高温活性中心数量而导致催化活性下降。XPS表征结果显示,La的加入可有效抑制Sn的还原,使其稳定在氧化态以维持催化剂的活性。TPO表征结果显示,La可有效避免催化剂结焦,但当La过量时,积碳量也会相应增加。Yu等[23]在PtSn/γ-Al2O3催化剂中添加元素Ce修饰载体γ-Al2O3,发现Ce的加入在很大程度上改善了催化剂的脱氢性能和稳定性,在576 ℃、空速3 800 h-1的条件下反应50 h后,丙烷转化率仍大于38%,丙烯选择性大于98%,丙烯收率为37%。他们认为:Ce的加入不仅保持了Pt催化剂的活性位,而且抑制了积碳在催化剂表面的聚集。Pisduangdaw等[24]采用火焰喷射热分解法制备了PtSnCe/Al2O3催化剂,并研究了该催化剂在丙烷脱氢反应中的催化性能,发现Ce的加入可提高Pt 的分散度、催化剂的活性和稳定性。
总之,在Pt系催化剂中适量添加助剂,对催化剂活性组分的分散度以及催化剂的酸碱性、活性、稳定性和选择性均具有明显的促进作用;此外,还可抑制副反应的发生、提高催化剂的抗积碳性能。目前在稀土金属助剂方面的研究相对较少,可在这方面加大研究力度。
2.4 载体
在负载型催化剂中,载体的主要作用:1) 与活性组分形成新的化合物,发挥特有的复合功能;2) 增加有效比表面积,提供合适的孔结构;3)提高催化剂的机械强度;4) 改善催化剂的热稳定性;5) 提供活性中心;6) 节省活性组分用量、降低成本;7) 增强催化剂的抗中毒能力;8) 均相催化剂负载化。
目前,在丙烷无氧脱氢催化体系中,用于Pt系催化剂的载体主要为γ-Al2O3。γ-Al2O3由于具有发达的孔隙结构、大的比表面积、适宜的表面性质、优异的热稳定性及机械强度而被广泛用作金属和金属氧化物催化剂的载体。对其他种类的氧化物及分子筛也有研究,如SiO2、SAPO-34、SBA-15、ZSM-5、MCM-41、尖晶石MgAl2O4和ZnAl2O4等,这些载体由于热稳定性和机械强度高、表面酸性低,适合低碳烷烃在苛刻条件下脱氢,也可作为丙烷脱氢催化剂的载体。董文生[25]研究了不同载体(Al2O3,MgAl2O4,ZnAl2O4)负载PtSn催化剂的丙烷脱氢反应性能。实验结果表明,在550 ℃下丙烷脱氢活性高低的顺序为:Pt/Al2O3>Pt/ZnAl2O4>Pt/MgAl2O4,丙烯选择性高低的顺序为:Pt/ZnAl2O4>Pt/Al2O3>Pt/MgAl2O4。催化性能的差异主要取决于:1)Pt与载体之间相互作用的不同;2)不同载体具有不同的表面性质。王秀玲[26]在研究分子筛载体对Pt-Sn-Na三组分丙烷脱氢催化剂性能的影响时发现,不同比例的SiO2和Al2O3负载三组分丙烷脱氢催化剂的性能不同,其中以n(SiO2)∶n(Al2O3)=100的ZSM-5分子筛和n(SiO2)∶n(Al2O3)=80的β分子筛进行机械混合得到的混合分子筛为载体制备的Pt-Sn-Na/ZSM-5-β催化剂的丙烷脱氢性能较好,在反应温度600 ℃、压力0.1 MPa时,丙烯选择性大于等于98.6%。她认为这是因为两种载体之间存在着协同作用。Kumar等[27]研究了Pt负载于ZSM-5沸石、Beta沸石和SBA-15分子筛上的丙烷脱氢性能。实验结果表明,丙烯选择性高低的顺序为:Pt/ZSM-5>Pt/Beta>Pt/SBA-15,表明催化剂的活性随载体尺寸的增大而降低;结焦量大小的顺序为:Pt/ZSM-5>Pt/SBA-15>Pt/Beta,表明Pt/Beta催化剂比Pt/ZSM-5和Pt/SBA-15催化剂具有更优的抗积碳性能。Pt/ZSM-5催化剂虽结焦量最大,但其选择性和稳定性最高,说明微孔载体比介孔载体具有更佳的催化性能。
Wang等[28]研究了ZnO改性MgAl2O4载体负载PtSn催化剂对丙烷脱氢反应的催化性能。实验结果表明,载体经改性后,催化剂的活性、稳定性及丙烯的选择性均优于传统的PtSn/Al2O3催化剂。 他们认为,这是因为经ZnO改性后,金属Pt的分散度增大,同时加强了金属Pt与载体之间的相互作用。
2.5 催化剂的制备方法
传统的催化剂制备方法有机械混合法、沉淀法、浸渍法、溶液蒸干法、热熔融法、浸溶法(沥滤法)和离子交换法等。余长林[29]研究了制备方法对丙烷无氧脱氢PtSn/γ-Al2O3催化剂性能的影响,发现制备方法对活性组分在催化剂表面的分散状态及Sn组分的存在状态有重要影响。黄宁表等[30]合成了3种Pt-Sn原子簇合物Pt(SnCl3)(PPh3)2,(Me4N)2[Pt(SnCl3)2Cl2],(Me4N)3[Pt(SnCl3)5],分别以这3种原子簇合物的有机溶液浸渍γ-Al2O3载体,制得3种负载型PtSn原子簇催化剂,并与传统浸渍法制备的负载型PtSn催化剂进行对比,发现前者比后者具有更高的催化活性、稳定性和选择性。这表明通过原子簇制备PtSn催化剂是提高PtSn催化剂性能的有效途径。盖希坤[31]分别用等体积浸渍法和喷涂法制备了PtSn/γ-Al2O3催化剂并研究了它们的催化性能。实验结果表明,等体积浸渍法制备的催化剂的丙烯收率和选择性均比喷涂法制备的催化剂高,且丙烯收率和选择性均比较稳定。这与美国专利[32]中阐述的喷涂法制得的催化剂的效果好于浸渍法制得的催化剂有所矛盾,究其原因可能是前者在制备过程中溶液喷涂不均匀所致。
Kappenstein等[33]采用共浸渍法或以[Pt(NH3)4]·[SnCl6]为前体制备了PtSn/Al2O3催化剂,并研究了这两种方法制备的催化剂对环己烷脱氢反应的催化性能。研究结果表明,以[Pt(NH3)4][SnCl6]为前体制备的催化剂的活性低于采用共浸渍法制备的催化剂。其原因是:用前体制备的催化剂中紧密相邻的Pt原子簇的个数少,而环己烷脱氢反应需较多紧密相邻的Pt原子簇。
3 催化剂的失活
Pt系催化剂的失活主要由积碳和Pt粒子烧结引起。积碳是烷烃分子在负载型金属催化剂表面深度脱氢和在载体酸性表面聚合、环化过程形成的含碳沉积物。催化剂表面积碳包括金属表面积碳和载体表面积碳[33],二者的差异在于前者的H/C比高,而后者的H/C比低,且后者对催化剂活性的影响较小。Pt粒子烧结主要是高温下活性组分小晶粒Pt具有较大的自由能,表面晶格质点热振动产生位移,逐渐由小晶粒Pt聚集为大晶粒Pt,导致Pt的活性表面减小,从而降低催化剂的活性和选择性[12]。
催化剂表面积碳失活可能由多种因素引起,其中催化剂的组成、结构和性质是影响积碳的首要因素[34]。Lin等[35]研究了PtSn/Al2O3催化剂表面的积碳行为,认为积碳是动态过程,烷烃分子在金属表面经深度脱氢形成积碳前体沉积在金属表面或从金属表面迁移至载体上,Sn组分的加入可降低积碳前体深度脱氢程度,促进积碳前体由金属表面迁移至载体,增强催化剂的抗积碳能力,使之具有更高的活性及稳定性。
Pt晶粒烧结是导致催化剂失活的另一个重要原因。Fiedorow等[36]在研究负载型催化剂失活时发现,在550 ℃以下,Pt晶粒表面部分Pt原子被氧化成PtO2,而PtO2在载体表面迁移时被载体表面高能位置捕获的机会大于其团聚的机会,故氧化使Pt再分散;当温度超过550 ℃时,迁移物种相互聚集形成大的晶粒,即Pt晶粒烧结造成催化剂失活。 Lin等[37]研究了高分散双组分催化剂的表面结构并将之应用于异丙烷脱氢反应,根据催化剂活性评价及H2吸附表征结果得出,晶粒烧结是Pt/Al2O3催化剂失活的主要因素。李庆[38]在研究Pt催化剂上丙烷脱氢反应与结焦动力学时分析了Pt催化剂上的结焦机理(见图4)。丙烷首先在金属上发生解离吸附,吸附的丙基经脱氢生成焦碳前体,该焦碳前体一部分生成金属上的焦,另一部分可迁移至载体,参与载体上焦碳的生成。此外,丙烯可直接吸附在载体的酸性位上,参与载体上的结焦过程。
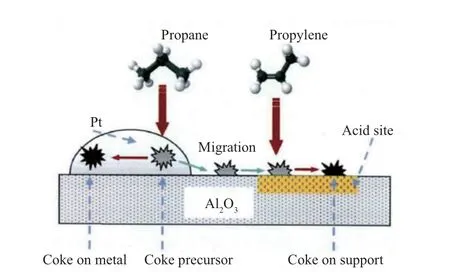
图4 Pt催化剂上的结焦机理Fig.4 Coke formation mechanism on a Ptcatalyst.
董文生等[10]指出,PtSn/Al2O3催化剂催化丙烷脱氢过程中,生成的副产物主要有甲烷、乙烯、乙烷和碳,丙烷脱氢生成丙烯和丙烷裂解生成甲烷、乙烯同时进行,且乙烯比丙烯生成的速率更快。他们认为,乙烯是催化剂积碳的前体,而甲烷只是与丙烯竞争生成的副产物。
工业上使用的Cr2O3/Al2O3催化剂由于经常处于氧化、还原态,使其物化性质发生改变从而导致催化剂寿命缩短。Puurunen等[39]指出,Cr2O3/Al2O3催化剂的可再生失活可能是因为:1)高温下Cr活性中心烧结;2)Al2O3载体的酸性促进了积碳的形成,生成的积碳覆盖了Cr活性中心。而Cr2O3/Al2O3催化剂的永久失活是由于生成了Cr3+物种。
4 结语
丙烷无氧脱氢制丙烯技术是解决丙烯持续增长需要的一条重要途径,具有可观的应用前景,目前已受到广泛的关注。但对于该反应及催化体系的许多理论问题还亟待解决,如反应机理、反应动力学、助剂的作用、如何获得适合的载体、如何提高催化剂的稳定性等。这些问题的深入研究将对进一步改进催化剂的性能及优化操作条件提供重要指导。
丙烷无氧脱氢催化剂主要为Pt系和Cr系催化剂。其中,Pt系催化剂具有高的脱氢活性,但在高温下Pt颗粒易烧结而导致活性中心数量减少、裂解和积碳反应加剧、丙烯选择性下降。因此,Pt系催化剂对丙烷无氧脱氢反应的选择性和稳定性还有较大的提升空间。目前对Pt系催化剂进行改性的主要途径有加入助剂、改变催化剂载体和催化剂的制备方法等。目前在工业上使用的大多仍为Cr系催化剂,但它失活较快、再生频繁、经济性低、有毒,使其广泛应用受到限制,因此开发低Cr含量的新型催化剂是关键;此外,Cr系催化剂上丙烷脱氢机理及反应动力学也有待进一步研究。
[1]Zangeneh F T,Sahebdelfar S,Bahmani M. Propane Dehydrogenation over a Commercial Pt-Sn/Al2O3Catalyst for Isobutane Dehydrogenation:Optimization of Reaction Conditions[J].Chin J Chem Eng,2013,21(7):730 - 735.
[2]Sahebdelfar S,Ravanchi M T,Zangeneh F T,et al. Kinetic Study of Propane Dehydrogenation and Side Reactions over Pt-Sn/Al2O3Catalyst[J]. Chem Eng Res Des,2012,90(8):1090 - 1097.
[3]Zhang Yiwei,Zhou Yuming,Shi Junjun,et al. Propane Dehydrogenation over PtSnNa/La-Doped Al2O3Catalyst:Effect of La Content[J]. Fuel Process Technol,2013,111:94 - 104.
[4]Bai Linyang,Zhou Yuming,Zhang Yiwei,et al. Inf l uence of the Competitive Adsorbates on the Catalytic Properties of PtSn-NaMg/ZSM-5 Catalysts for Propane Dehydrogenation[J]. Ind Eng Chem Res,2011,50(8):4345 - 4350.
[5]陈建九,史海英,汪泳. 丙烷脱氢制丙烯工艺技术[J]. 精细石油化工进展,2000(12):23 - 28.
[6]马艳萍,杨茹欣,赵燕. 丙烷催化脱氢制丙烯生产技术及工业应用[J]. 广东化工,2012,39(7):87,115.
[7]吴锁林. 丙烷脱氢制丙烯的技术进展[J].江苏化工,1998,26(2):33 - 35.
[8]吴泽彪,杨维慎,李吴义,等. 无机膜反应器中丙烷脱氢制丙烯的数学模型[J]. 催化学报,1997,18(1):38 - 41.
[9]Kah Sing Ho,Joanna Jo Ean Chye,Sim Yee Chin,et al.Characterization of Industrial Pt-Sn/Al2O3Catalyst and Transient Product Formations During Propane Dehydrogenation[J].Bull Chem React Eng Catal,2013,8(1):77 - 82.
[10]董文生,王心葵,彭少逸 . 丙烷脱氢制丙烯研究进展[J].合成化学,1997,5(3):246 - 250.
[11]Cimino A,Cordishi D,Derossi S. Studies on Chromia Zirconia Catalysts:Ⅲ. Propylene Hydrogenation[J]. J Catal,1991,12(7):777 - 787.
[12]余长林,葛庆杰,徐恒泳,等. 丙烷脱氢制丙烯研究新进展[J]. 化工进展,2006,25(9):977 - 983.
[13]Benesl H A,Curtis R M. Preparation of Highly Dispersed Catalytic Metals:Platinum Supported on Silica Gel[J]. J Catal,1968,10(4):328 - 335.
[14]Biloen P,Dautzenberg,Sachtler W M H. Catalytic Dehydrogenation of Propane to Propene over Platinum and Platinum-Gold Alloys[J]. J Catal,1977,50(1):77 - 86.
[15]Somorjai G A,Li Yimin. Introduction to Surface Chemistry and Catalysis[M]. New York:Wiley-Interscience,2010:21 - 22.
[16]Kumar M S,Chen De,Walmsle J C,et al. Dehydrogenation of Propane over Pt-SBA-15:Effect of Pt Particle Size[J].Catal Commun,2008,9(5):747 - 750.
[17]Kumar M S,Chen De,Anderson H,et al. Dehydrogenation of Propane over Pt-SBA-15 and Pt-Sn-SBA-15:Effect of Sn on the Dispersion of Pt and Catalytic Behavior[J]. Catal Today,2009,142(1/2):17 - 23.
[18]Zeeshan Nawaz,Tang Xiaoping,Zhang Qiang,et al. SAPO-34 Supported Pt-Sn-Based Novel Catalyst for Propane Dehydrogenation to Propylene[J]. Catal Commun,2009,10(14):1925 - 1930.
[19]Zhang Yiwei,Zhou Yuming,Qiu Anding,et al. Propane Dehydrogenation on PtSn/ZSM-5 Catalyst:Effect of Tin as a Promoter[J]. Catal Commun,2006,7(11):860 - 866.
[20]余长林,葛庆杰,徐恒泳,等. 助剂对Pt/γ-Al2O3催化剂丙烷脱氢性能的影响[J]. 石油化工,2006,35(3):217 -220.
[21]Huang Li,Zhou Shijie,Zhou Yuming,et al. Effect of Strontium Addition to Platinum Catalyst for Propane Dehydrogenation[J]. China Pet Process Pe,2012,14(3):75 - 82.
[22]张一卫,周钰明,叶志良,等. 镧对PtSnNa/Al2O3催化剂丙烷脱氢反应性能的影响[J]. 现代化工,2006,26(2):33 - 38.
[23]Yu Changlin,Ge Qingjie,Xu Hengyong,et al. Effects of Ce Addition on the Pt-Sn/γ-Al2O3Catalyst for Propane Dehydrogenation to Propylene[J]. Appl Catal,A,2006,315:58 - 67.
[24]Pisduangdaw S,Panpranot J,Chaisuk C,et al.Flame Sprayed Trimetallic Pt-Sn-X/Al2O3Catalysts(X=Ce,Zn,and K)for Propane Dehydrogenation[J]. Catal Commun,2011,12(12):1161 - 1165.
[25]董文生. 不同载体的PtSn催化剂上丙烷脱氢性能的研究[J].天然气化工,1999,24(3):9 - 12.
[26]王秀玲. 分子筛载体对Pt-Sn-Na三组分丙烷脱氢催化剂性能的影响[J].石油化工,2012,41(12):1346 - 1350.
[27]Kumar M S,Holmen A,Chen De. The Inf l uence of Pore Geometry of Pt Containing ZSM-5,Beta and SBA-15 Catalysts on Dehydrogenation of Propane[J]. Microporous Mesoporous Mater,2009,126(1/2):152 - 158.
[28]Wang Yaojie,Wang Yanmei,Wang Shurong,et al. Propane Dehydrogenation over PtSn Supported on ZnO-Modified MgAl2O4[J]. Catal Lett,2009,132(3/4):472 - 479.
[29]余长林. 丙烷脱氢铂催化剂与反应性能的研究[D]. 大连:中国科学院大连化学物理研究所,2007.
[30]黄宁表,田金忠,刘强,等. 铂锡双金属催化剂上丙烷脱氢反应研究[J]. 燃料化学学报,1996,24(2):108 - 113.
[31]盖希坤. 丙烷催化脱氢制丙烯的研究[D]. 青岛:山东科技大学,2008.
[32]Air Products and Chemical Shic. Catalytic Dehydrogenation Reactor Cycle:US,4581339 A[P]. 1986-04-08.
[33]Kappenstein C,Gue′rin M,Lázár K,et al. Characterisation and Activity in n-Hexane Rearrangement Reactions of Metallic Phases on Pt-Sn/Al2O3Catalysts of Different Preparations[J].J Chem Soc,Faraday Trans,1998,94(16):2463 - 2473.
[34]张涛,臧憬龄,胡爱华,等. 锂对于γ-Al2O3表面酸中心的跳板作用及其积炭性能的影响[J]. 催化学报,1990,11(5):341 - 347.
[35]Lin Liwu,Zhang Tao,Zang Jinglin,et al. Dynamic Process of Carbon Deposition on Pt and Pt-Sn Catalysts for Alkane Dehydrogenation[J]. Appl Catal,1990,67(1):11 - 23.
[36]Fiedorow R M J,Chahar B S,Wanke S E. The Sintering of Supported Metal Catalysts:Ⅱ. Comparison of Sintering Rates of Supported Pt,Ir,and Rh Catalysts in Hydrogen and Oxygen[J]. J Catal,1978,51(2):193 - 202.
[37]Lin Liwu,Yang Weisheng,Jia Jifei. Surface Structure and Reaction Performances of Highly Dispersed and Supported Bimetallic Catalysts[J]. Sci China,Ser B,1999,42(6):571 - 580.
[38]李庆. Pt催化剂上丙烷脱氢反应与结焦动力学[D]. 上海:华东理工大学,2012.
[39]Puurunen R L,Weckhuysen B M. Spectroscopic Study on the Irreversible Deactivation of Chromia/Alumina Dehydrogenation Catalysts[J]. J Catal,2002,210:418 - 430.
