p38MAPK在心力衰竭发病机制中的研究进展
2014-03-28王淑香综述审校
王淑香(综述),赵 强(审校)
(暨南大学第四附属医院 广州市红十字会医院心血管内科,广州 510220)
促分裂原活化的蛋白激酶(mitogen-activated protein kinase,MAPK)属于丝氨酸或者苏氨酸蛋白激酶,它可以被某些配体包括受体、生长因子、G蛋白偶联以及一些应激源所激活。p38MAPK与细胞外信号调节酶1/2、细胞外调节信号5以及c-Jun N端激酶(c-Jun N-terminal kinase,JNK)同属于MAPK系统,并可以被应激源所激活。JNK、p38MAPK、细胞外调节信号5可在人类心脏中表达,JNK、p38MAPK的活性可在缺血性心肌病的心力衰竭(心衰)中明显升高[1]。p38MAPK、细胞外调节信号5则可选择性地在小鼠远期及心肌梗死后的心肌细胞中显著表达[2]。此后,相继有研究证实p38MAPK的激活还参与大鼠、犬、兔子以及某些鱼类的心衰的发展[3-6]。目前已知的主要机制包括对濒死心肌细胞的挽救与保护、心脏的重构、心脏功能的紊乱、炎性反应、氧化应激、内皮功能紊乱、细胞凋亡等。
1 p38MAPK与心脏结构重构、电重构
心脏重构是心衰发生与发展的基本过程,它包括在心腔扩大、心室肥厚的过程中,与心肌细胞、细胞外基质、胶原纤维网等各种组织结构改变相关的结构重构以及与各种信号通道改变相关的电重构。研究表明,p38MAPK在心衰发展过程中可被明显激活,并参与心室重构的过程,而p38MAPK的抑制对改善心室重构是有益的[2-3]。Lei等[3]用结扎左前降支动脉方法建立心肌梗死模型,并以心肌内注射药物的方法处理斯普拉格-杜勒鼠,通过对重组腺病毒携带的多配体聚糖1(syndecan-1,Sdc-1)互补DNA转染的大鼠研究发现,Sdc-1的过度表达可明显改善大鼠心肌梗死后出现的纤维化性的心脏重构,而在心脏重构的改善过程中,p38MAPK的活性受到了显著的抑制,提示p38MAPK的激活在诱导心脏重构纤维化过程中充当了积极角色,而这种作用是受Sdc-1调控的,p38MAPK的抑制可改善心脏重构过程。除了Sdc-1外,近年研究中发现参与此抑制p38MAPK过程的分子还包括激酶锚定蛋白-Lbc、微RNA-350以及心脏保护基因Dusp1和Dusp4[7-9]。Pérez等[7]在转基因小鼠的研究中发现,主动脉缩窄诱导的血压升高的反应中,激酶锚定蛋白-Lbc/p38-MAPK的混合抑制的破坏可以抑制代偿性心肌肥厚同时促进早期心功能紊乱,而这种心功能紊乱是与细胞凋亡、压力基因的激活以及心室扩大密切相关的。Ge等[8]发现,p38MAPK的抑制同样发生在微RNA-350诱导的大鼠病理性心肌肥厚的过程中,而其抑制现象与心脏保护基因Dusp1和Dusp4密切相关[9]。除心脏结构重构外,近年来在心脏电重构的研究上也有较为新颖的发现。Tang等[10]在对大鼠心肌梗死后6~8周的心室组织研究发现,左心室电压门控钾离子信号通道的表达是由氧化还原反应所调控,且心肌梗死后心脏硫氧还蛋白系统的慢性损害可通过持续激活的细胞凋亡信号调控的激酶1-JNK-p38MAPK的信号通道来诱导瞬时外向钾电流的重构,提示心脏的硫氧还蛋白系统可能为心衰中逆转或防止心律失常的重要靶点。
2 p38MAPK与心肌纤维化
心肌纤维化是胶原在心肌细胞外基质过量沉积的结果,是心脏疾病中重要而常见的病理过程,它在心脏功能从代偿到失代偿转变、心肌重构从可逆到不可逆变化中发挥至关重要的作用。心肌纤维化及细胞凋亡均是心衰的早期特征[11]。较早前,Barasch等[12]通过嵌入式的病例对照分析方法,对869例平均年龄77岁且男女比例相当的老年患者(包括131例收缩性心衰患者,179例舒张性心衰患者,280例具有心血管高危因素者以及279例健康人)的血清纤维化指标进行检测发现,纤维化的标志物Ⅰ型胶原、Ⅲ型前胶原在舒张性或收缩性心衰的老年患者中均显著升高,两者并无统计学意义,提示心肌纤维化在心衰的发展过程中与年龄的增长密切相关。
心肌纤维化在分子水平的机制十分复杂,包括众多分子及细胞成分的参与,如近年文献中提到的骨髓来源的细胞、胰岛素样生长因子、蛋白激酶C(protein kinase C,PKC)β2以及p38MAPK[13-15]。骨髓来源的细胞在基质导出因子1的影响下可促进慢性心衰患者心脏的心肌纤维化[13]。胰岛素样生长因子1可通过阻滞心肌纤维化以及血清应答因子依赖的结缔组织生长因子诱导的途径来改善心脏功能[14]。PKCβ2可促进心衰进展,持续选择性抑制PKCβ2同样可改善心肌梗死后心衰的心脏功能,而这与心肌纤维化以及前炎性反应的减少密切相关[15]。众多实验资料证明,p38MAPK的激活参与了心肌纤维化过程[16-19]。2007年,Cortez等[16]在人类主要的成纤维细胞中的研究发现,白细胞介素(interleukin,IL)17诱导的基质金属蛋白酶(matrix metalloproteinase,MMP)1通过p38MAPK以及细胞外调节信号通道依赖的激活蛋白1进行表达。近年研究发现,p38MAPK的激活同样参与了人类成纤维细胞中细胞因子诱导的IL-6、MMP-3,肿瘤坏死因子α诱导的MMP-9的表达以及在低氧状态下成年大鼠心肌成纤维细胞中低氧诱导的G0/G1阻滞[17-19]。然而,它们之间的相互作用关系目前还没有完全明确。
3 p38MAPK与β肾上腺素能受体
近年报道了一些关于p38MAPK与β肾上腺素能受体(β-adrenergic receptor,β-AR),包括:β1-AR、β2-AR、β3-AR等在心衰中作用机制的研究相关的文献,并揭示了两者在心衰发展过程中可能充当的角色,见图1。其中以β2-AR的研究居多。Xu等[20]通过对转基因β2-AR超量表达的小鼠进行研究发现,β2-AR的激活可刺激烟酰胺腺嘌呤二核苷酸磷酸氧化酶衍生的活性氧类(reactive oxygen species,ROS)增多,而增多的ROS可激活p38MAPK导致明显的心脏炎性反应、心脏重构以及心衰。然而,关于ROS与p38MAPK的上下游关系目前尚存在争议。有学者认为,p38MAPK才是ROS的上游调节,ROS的激活可通过肌纤维的氧化影响着左心室的功能[4]。另一研究中则指出,β2-AR的过度表达可通过减少烟酰胺腺嘌呤二核苷酸磷酸2/4氧化酶的上调导致ROS的释放增多,而ROS的增多同时伴随着p38MAPK活性的增高、心肌的纤维化、细胞凋亡以及心脏功能的紊乱[21]。除β2-AR外,近年也有关于β1-AR与p38MAPK在心衰作用中的研究。2012年Chen等[22]研究发现,环磷酸腺苷直接激活交换蛋白依赖激活的p38MAPK和PKC共同参与了在新生老鼠心脏纤维细胞中β-AR激活的IL-6生成的过程。同年Du等[23]采用与正常人对照的方法,对95例扩张型心肌病患者的血液样本分析发现,β1-AR自身抗体可通过β1-AR/cAMP/PKA和p38MAPK激活途径促使T淋巴细胞增生,这揭示了p38MAPK与β1-AR两者参与心衰发展的免疫途径。目前对于β3-AR在心衰中的作用研究相对较少,近年有学者采用腹主动脉缩窄方法建立大鼠心衰模型中发现,β3-AR在心衰大鼠心脏的心房及心室均显著升高[24-25]。另有研究证实,β3-AR在小鼠超压力负荷的肥大心肌以及心衰中能起到保护心脏作用[26],对幼年及成年的虹鳟鱼的心血管具有调控作用[5]。至于p38MAPK与β3-AR以及其他β-AR在心衰中的作用机制仍待进一步研究。
4 小 结
纵观近年p38MAPK在心衰发生机制中的研究,主要集中在心室重构、心肌纤维化以及氧化应激等方面。随着时间的推移,p38MAPK在心衰发生中所担任的角色将日渐明朗。目前关于 p38MAPK 各种抑制剂的实验也不断增多,研究水平也在不断深入。但由于其作用机制复杂,除上述几个近年较受欢迎的研究点外,其他如炎症反应、免疫应答、细胞保护等机制的研究仍十分欠缺。纵使在热门的机制研究点上,其相关分子的作用链仍未完全清楚。将来如何在已认识的机制中拓展研究,寻找潜在的作用机制以及更有价值分子靶向点,为心衰治疗的发展迈上新一步,会是留给人类科学研究最大的难题。
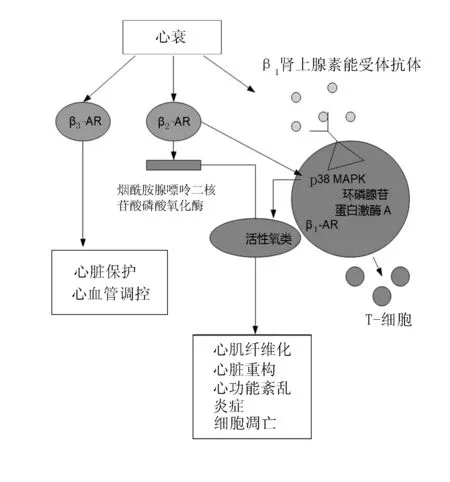
p38AMPK:促分裂原活化的p38的蛋白激酶
[1] Cook SA,Sugden PH,Clerk A.Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease[J].J Mol Cell Cardiol,1999,31(8):1429-1434.
[2] Yeh CC,Li H,Malhotra D,etal.Distinctive ERK and p38 signaling in remote and infarcted myocardium during post-MI remodeling in the mouse[J].J Cell Biochem,2010,109(6):1185-1191.
[3] Lei J,Xue S,Wu W,etal.Sdc1 overexpression inhibits the p38 MAPK pathway and lessens fibrotic ventricular remodeling in MI rats[J].Inflammation,2013,36(3):603-615.
[4] Heusch P,Canton M,Aker S,etal.The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits[J].Br J Pharmacol,2010,160(6):1408-1416.
[5] Petersen LH,Needham SL,Burleson ML,etal.Involvement of β(3)-adrenergic receptors in in vivo cardiovascular regulation in rainbow trout (Oncorhynchus mykiss)[J].Comp Biochem Physiol A Mol Integr Physiol,2013,164(2):291-300.
[6] Sharov VG,Todor A,Suzuki G,etal.Hypoxia,angiotensin-II,and norepinephrine mediated apoptosis is stimulus specific in canine failed cardiomyocytes:a role for p38 MAPK,Fas-L and cyclin D1[J].Eur J Heart Fail,2003,5(2):121-129.
[7] Pérez López I,Cariolato L,Maric D,etal.A-kinase anchoring protein Lbc coordinates a p38 activating signaling complex controlling compensatory cardiac hypertrophy[J].Mol Cell Biol,2013,33(15):2903-2917.
[8] Ge Y,Pan S,Guan D,etal.MicroRNA-350 induces pathological heart hypertrophy by repressing both p38 and JNK pathways[J].Biochim Biophy,2013,1832(1):1-10.
[9] Auger-Messier M,Accornero F,Goonasekera SA,etal.Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy[J].Cric Res,2013,112(1):48-56.
[10] Tang K,Li X,Zheng MQ,etal.Role of apoptosis signal-regulating kinase-1-c-Jun NH2-terminal kinase-p38 signaling in voltage-gated K+channel remodeling of the failing heart:regulation by thioredoxin[J].Antioxid Redox Signal,2011,14(1):25-35.
[11] Gürtl B,Kratky D,Guelly C,etal.Apoptosis and fibrosis are early features of heart failure in an animal model of metabolic cardiomyopathy[J].Int J Exp Pathol,2009,90(3):338-346.
[12] Barasch E,Gottdiener JS,Aurigemma G,etal.Association between elevated fibrosis markers and heart failure in the elderly:the cardiovascular health study[J].Circ Heart Fail,2009,2(4):303-310.
[13] Chu PY,Mariani J,Finch S,etal.Bone marrow-derived cells contribute to fibrosis in the chronically failing heart[J].Am J Pathol,2010,176(4):1735-1742.
[14] Touvron M,Escoubet B,Mericskay M,etal.Locally expressed IGF1 propeptide improves mouse heart function in induced dilated cardiomyopathy by blocking myocardial fibrosis and SRF-dependent CTGF induction[J].Dis Model Mech,2012,5(4):481-491.
[15] Palaniyandi SS,Ferreira JC,Brum PC,etal.PKCβII inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses[J].J Cell Mol Med,2011,15(8):1769-1777.
[16] Cortez DM,Feldman MD,Mummidi S,etal.IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK-and ERK1/2-dependent C/EBP-beta,NF-kappaB,and AP-1 activation[J].Am J Physiol Heart Circ Physiol,2007,293(6):h3356-3365.
[17] Sinfield JK,Das A,O′Regan DJ,etal.p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts[J].Biochem Biophys Res Commun,2013,430(1):419-424.
[18] Yang CM,Lee IT,Lin CC,etal.c-Src-dependent MAPKs/AP-1 activation is involved in TNF-α-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells[J].Biochem Pharmacol,2013,85(8):1115-1123.
[19] Pillai MS,Sapna S,Shivakumar K.p38 MAPK regulates G1-S transition in hypoxic cardiac fibroblasts[J].Int J Biochem Cell Biol,2011,43(6):919-927.
[20] Xu Q,Dalic A,Fang L,etal.Myocardial oxidative stress contributes to transgenic β-adrenoceptor activation-induced cardiomyopathy and heart failure[J].Br J Pharmacol,2011,162(5):1012-1028.
[21] Di Lisa F,Kaludercic N,Paolocci N.β-Adrenoceptors,NADPH oxidase,ROS and p38 MAPK:another ‘radical’ road to heart failure?[J].Br J Pharmacol,2011,162(5):1009-1011.
[22] Chen C,Du J,Feng W,etal.β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCδ/p38 MAPK signalling in neonatal mouse cardiac fibroblasts[J].Br J Pharmacol,2012,166(2):676-688.
[23] Du Y,Yan L,Wang J,etal.β1-Adrenoceptor autoantibodies from DCM patients enhance the proliferation of T lymphocytes through the β1-AR/cAMP/PKA and p38 MAPK pathways[J].PLoS One,2012,7(12):e52911.
[24] Zhao Q,Zeng F,Liu JB,etal.Upregulation of β3-adrenergic receptor expression in the atrium of rats with chronic heart failure[J].J Cardiovasc Pharmacol Ther,2013,18(2):133-137.
[25] Zhao Q,Wu TG,Jiang ZF,etal.Effect of beta-blockers on beta3-adrenoceptor expression in chronic heart failure[J].Cardiovasc Drugs Ther,2007,21(2):85-90
[26] Niu X,Watts VL,Cingolani OH,etal.Cardioprotective effect of beta-3 adrenergic receptor agonism:role of neuronal nitric oxide synthase[J].J Am Coll Cardiol,2012,59(22):1979-1987.
