超声波萃取-高效液相色谱-串联质谱同时测沉积物中10种PPCPs化合物
2014-03-24张盼伟赵高峰周怀东余丽琴李科林
张盼伟,赵高峰,周怀东,余丽琴,李 昆,文 武,李科林
1.中南林业科技大学环境科学与工程研究中心,湖南 长沙 410004
2.中国水利水电科学研究院,北京 100038
近年来,药物及个人护理产品(PPCPs)作为一类“新兴”的污染物已经受到人们的普遍关注。随着现代医学技术的发展,药物的销售量和使用量呈逐年上升趋势。在我国,药物(特别是抗生素类药物)滥用情况严重,有可能造成严重的环境污染。目前国内关于此类污染物的分析检测方法还不成熟,有关PPCPs污染的报道较少。国内外已经有学者开始对这一“新兴”的污染物进行研究[1-3],主要针对地表水[4-5]、地下水[6]、饮用水[7]、农业土壤[8-9]以及河流沉积物[10]中的PPCPs。在国内外使用高效液相色谱分离技术检测抗生素类化合物已有报道[11-15],但是在国内使用灵敏度高,定性、定量能力强的液相色谱-串联质谱(HPLC-MS/MS)技术同时检测沉积物中多种抗生素类化合物的报道较少。该研究采用HPLC-MS/MS,在EPA 1694方法[16]的基础上进行了改进,以期降低目标化合物的检测限,并且在保证目标化合物回收率效果的情况下尽可能地减少样品预处理时间,降低预处理成本。
1 实验部分
1.1试剂与材料
10种PPCPs分别是对乙酰氨基酚(acetaminophen, 纯度 99.5%)、林可霉素(lincomycin, 纯度 99.0%)、甲氧苄啶(trimethoprim, 纯度 99.5%)、咖啡因(caffeine, 纯度 98.5%)、阿奇霉素(azithromycin, 纯度 97.0%)、磺胺甲唑(sulfamethoxazole, 纯度 99.5%)、泰乐菌素(tylosin, 纯度 98.0%)、地尔硫卓(diltiazem, 纯度 99.0%)、卡马西平(carbamazepine, 纯度 99.5%)、氟西汀(fluoxetine,纯度 99.0%)标准品,氘代阿特拉津(atrazine d5, 纯度≥99.5%)为回收率指示物,均购自德国。上述标准品用乙腈或甲醇配制成质量浓度为1 500 mg/kg的标准溶液,用甲醇配制成储备液备用。所有标准溶液储存于-18 ℃。甲醇、乙腈、乙酸乙酯和二氯甲烷均为农残级(J.T.Baker, Phillipsburg, USA),甲酸(色谱纯,DIKMA),实验用水均为超纯水(经MilliQ系统纯化,电阻率为18. 3 MΩ·cm)。
1.2仪器与设备
Agilent 1290型高效液相色谱仪,配有Agilent 6460质谱检测器(ESI源);KQ-700DE型超声波清洗机,中国。CF16RXⅡ型离心机,日本;CHRIST冷冻干燥机,德国;Hei-VAP型,旋转浓缩仪,德国;N-EVAP-12型氮吹仪,美国。
1.3液相色谱和质谱条件
1.3.1色谱条件
ZORBAX Eclipse Plus C18反相柱(100 mm×2.1 mm,3.5 μm);流动相为0.1%(体积分数,下同)甲酸/水溶液(A),乙腈(B),流速为0.2 mL/min;梯度洗脱程序:8% B保持6 min,流速为0.2 mL/min,在10 min内由8% B增至30% B,流速从0.3 mL/min增至0.35 mL/min,随后在14 min内由30% B增至100% B;进样量10 μL,柱温30 ℃。
1.3.2质谱条件
电离源为电喷雾电离源(ESI),雾化器压力为310 kPa(45 psi),毛细管电压为4 000 V,干燥气温度为300 ℃,流速为9 L/min,检测方式为多反应监测(MRM)正离子模式。
1.4样品前处理方法
超声萃取:取4.0 g样品至50 mL的洁净三角锥瓶中,加入20 mL甲醇,30 ℃水浴中超声萃取(功率80%×200 W) 15 min。
离心净化:将上述上清液移入50 mL离心管中,以3 000 r/min的速度离心5 min,将上清液移至100 mL圆底烧瓶中。重复上述萃取步骤3次。将收集到的萃取液在气压350 hPa、水温50 ℃的条件下旋转浓缩至2~3 mL,待SPE净化。
SPE净化:SAX(500 mg, 3 mL)净化柱依次使用乙酸乙酯及二氯甲烷各5 mL清洗,10 mL甲醇浸泡30 s进行活化,萃取液通过净化柱后使用5 mL甲醇淋洗净化柱。
样品定容:将收集到的洗脱液在50 ℃水浴中使用缓和氮气吹干,使用0.1%(体积分数,下同)甲酸/甲醇溶液定容至500 μL,待HPLC-MS/MS检测。
2 结果与讨论
2.1色谱、质谱条件的选择和优化
根据EPA 1694方法,选取0.1%甲酸/水溶液(A)和乙腈(B)作为流动相。根据梯度洗脱[17-18],经过反复实验,最终确定了第1.3节所述梯度洗脱程序。在优化的梯度洗脱程序下,10种PPCPs以及氘代阿特拉津可进行较好分离(见图1)。

1.对乙酰氨基酚; 2.林可霉素;3.甲氧苄啶;4.咖啡因;5.阿奇霉素;6.磺胺甲唑;7.地尔硫卓;8.泰乐菌素;9.卡马西平;10.氟西汀;11.氘代阿特拉津
将10种PPCPs及氘代阿特拉津母液用甲醇配成工作液混标,首先进行全扫描(MS2-Scan模式),找出准确的[M+H]+分子离子峰,然后对其进行子离子扫描以获得二次碎裂产生的子离子。将分子离子和2个响应适中的子离子组成检测离子对,以MRM模式进行检测,在此基础上重点优化对灵敏度影响较大的碎裂电压和碰撞能量,使选定的母离子和子离子组成的特征离子的丰度和比例达到最佳。优化后参数见表1。
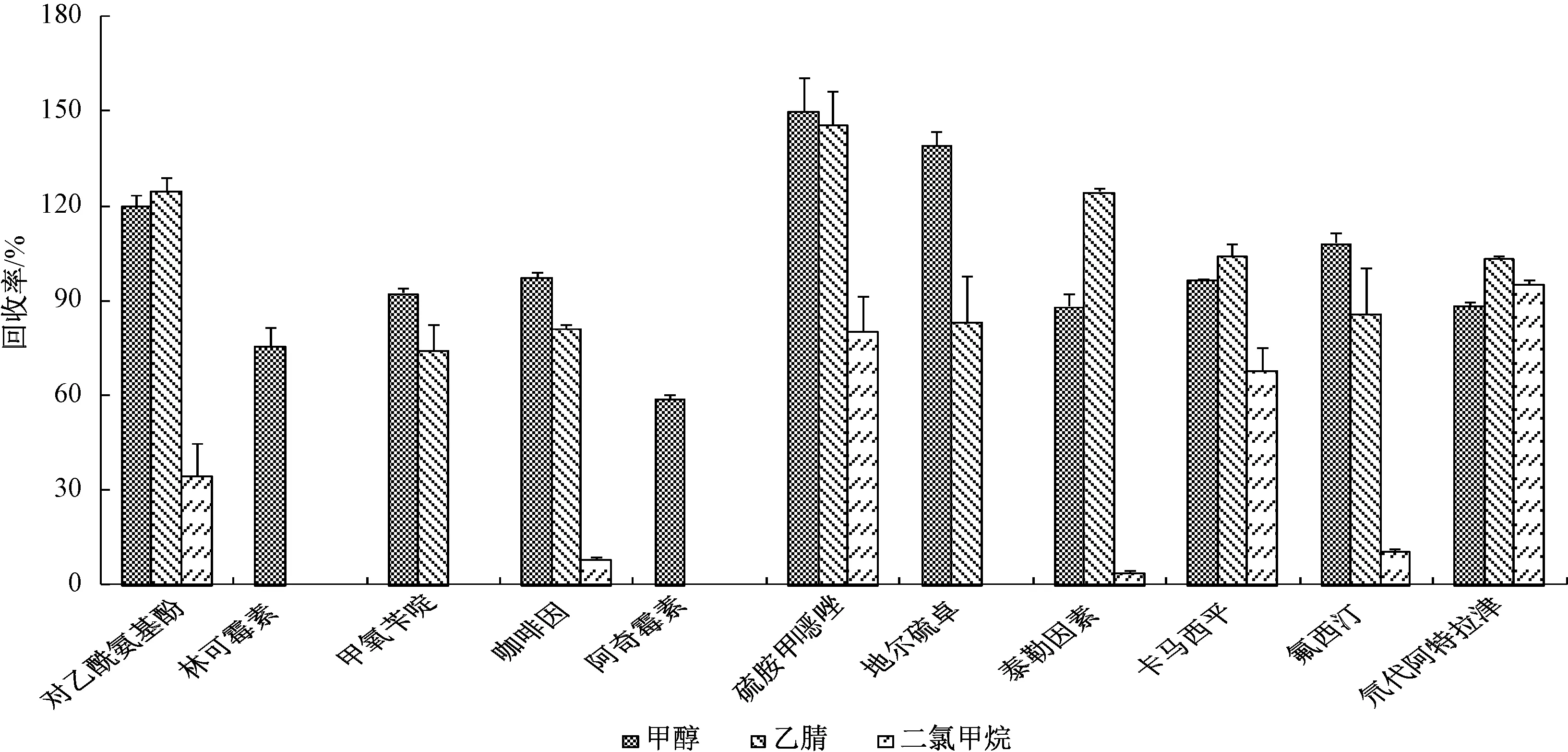
图2 3种萃取溶剂的回收率结果比较
表110种PPCPs化合物及回收率指示物的质谱条件

化合物m/z母离子子离子裂电压/V碰撞能量/eV对乙酰氨基酚152110 065.090901535林可霉素407 1359 1126.01101101525甲氧苄啶291261 0230.01101102525咖啡因195138 0110.01101101525阿奇霉素749 3375.2591 2158.01301303035磺胺甲唑254156 092.01101101525泰乐菌素916 3772 2174.01101103535地尔硫卓415178 0150.11301302525卡马西平237194 0179.01101101535氟西汀310148 11105氘代阿特拉津221179 0137.0101.0110110110151515
2.2固相萃取条件的选择和优化
2.2.1萃取溶剂的选择
选取EPA 1694方法及其他研究中提到的甲醇[19]、乙腈[16]和二氯甲烷[20-21]作为萃取溶剂进行回收率实验。实验根据萃取溶剂的不同分为3组,每组5个样品和1个实验空白。根据第1.4节所述步骤进行样品的前处理,并对其进行检测,3种萃取溶剂的回收率如图2所示。
从图2可以看出,甲醇对10种PPCPs及氘代阿特拉津的回收率都比较好,回收率范围为58.9%~154.0%,而乙腈和二氯甲烷对林可霉素和阿奇霉素的回收率几乎为零,二氯甲烷对甲氧苄啶、地尔硫卓等化合物的回收率也几乎为零,造成这种现象的原因可能是林可霉素与阿奇霉素较难溶于乙腈或二氯甲烷,甲氧苄啶与地尔硫卓较难溶于二氯甲烷,而甲醇对10种PPCPs及氘代阿特拉津都有很好的溶解作用。 因此,实验选取甲醇作为萃取溶剂。
2.2.2去色素填充柱的选择
实验选用填料为SAX的阴离子交换柱(500 mg,3 mL)及填料为PC/NH2(500 mg,6 mL)的净化柱,用乙酸乙酯和二氯甲烷各5 mL依次对净化柱进行清洗,然后用10 mL甲醇对柱子进行活化,将萃取液移入填充柱,并用5 mL的甲醇进行洗脱,回收率结果如图3所示。

图3 SAX净化柱与PC/NH2净化柱回收率结果
从图3可见, PC/NH2柱具有去除色素的作用,但是从回收率结果来看,PC/NH2柱除卡马西平及氘代阿特拉津的回收率较好外,对其他化合物的回收率较差。造成这种结果的原因可能是PC/NH2柱的填料对10种PPCPs化合物的吸附作用较强,化合物不容易被洗脱。而SAX阴离子交换柱效果较好,能有效去除样品中的色素,且10种PPCPs及氘代阿特拉津的回收率为77.6%~134.4%,方法简便,适合大批量样品的处理。因此,实验选取填料为SAX的阴离子交换柱。
2.2.3洗脱溶剂的用量选择
根据EPA 1694方法,选取甲醇作为SPE柱洗脱溶剂。每个样品分别收集前 5 mL与后5 mL洗脱液,最终定容至1 mL进行检测。从表2可以看出,前5 mL收集样品的回收率为69.33%~149.72%,后收集的5 mL样品中没有检测出目标化合物,说明5 mL的甲醇可以将目标化合物洗脱下来,且回收率较好。因此,实验选用溶剂用量为5 mL。回收率结果见表2。
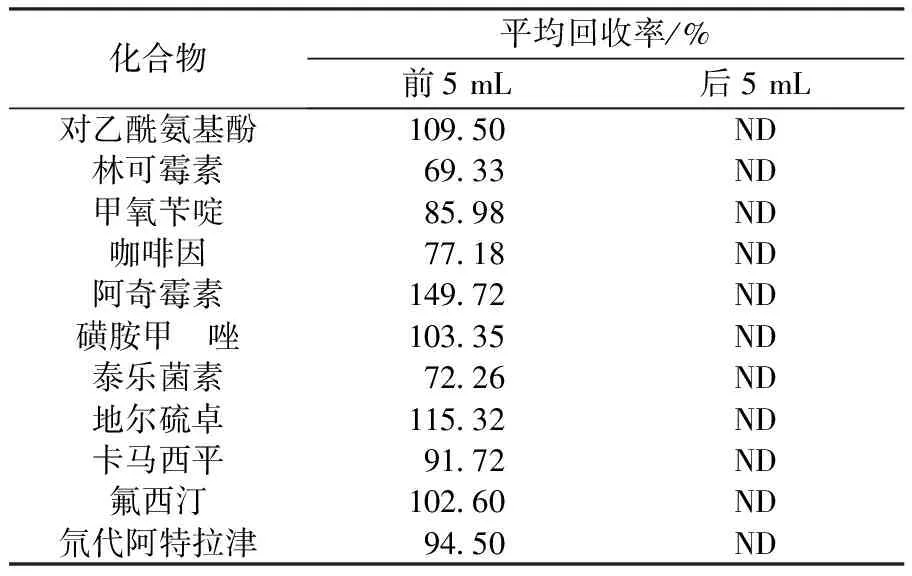
表 2 10种PPCPs化合物前5 mL与后5 mL洗脱液回收率结果(n=4)
2.3精密度及回收率实验
准确称取4.0 g石英砂作为样品基质,根据相关文献[22-24]报道中沉积物PPCPs的含量,选取2个加标浓度级别,分别为20、400 ng/g。根据第1.4节中的步骤对样品进行处理,每个浓度水平5个重复,用来考察方法的总体回收率和重现性;每个浓度水平添加2个空白样品用来检验实验过程是否会带来干扰。回收率指示物氘代阿特拉津在分析前加至石英砂中,对整个实验过程进行质量控制。10种PPCPs及氘代阿特拉津的回收率及RSD如表3所示。添加量为20 ng/g,回收率为61.1%~128.5%,RSD为1.7%~17.5%(n=5);添加量为400 ng/g,回收率为66.4%~126.7%,RSD为2.3%~18.0%(n=5)。该方法具有较好的重现性,精密度和回收率也符合要求。
2.4线性关系与检测限
配制0.1、1、10、50、100、200、500 ng/g 的标准溶液,根据子离子定量,10种PPCPs及氘代阿特拉津的线性相关系数r>0.99,方法的最低检测限以3倍信噪比进行统计,最低检测限为0.12~4.46 ng/g。线性关系及最低检测限见表3。
2.5实际沉积物样品中目标化合物分析
利用该文建立的方法对海河流域徒骇河和独流减河表层沉积物进行检测,结果见表4。可见,该方法可以去除土壤中的色素,并且添加的回收率指示物有较好的回收效果。在独流减河中有咖啡因(243.73 ng/g)的检出;而在徒骇河中有对乙酰氨基酚(0.42 ng/g)、林可霉素(55.78 ng/g)、咖啡因(1.56 ng/g)和阿奇霉素(10.75 ng/g)检出,以林可霉素为优势污染物。
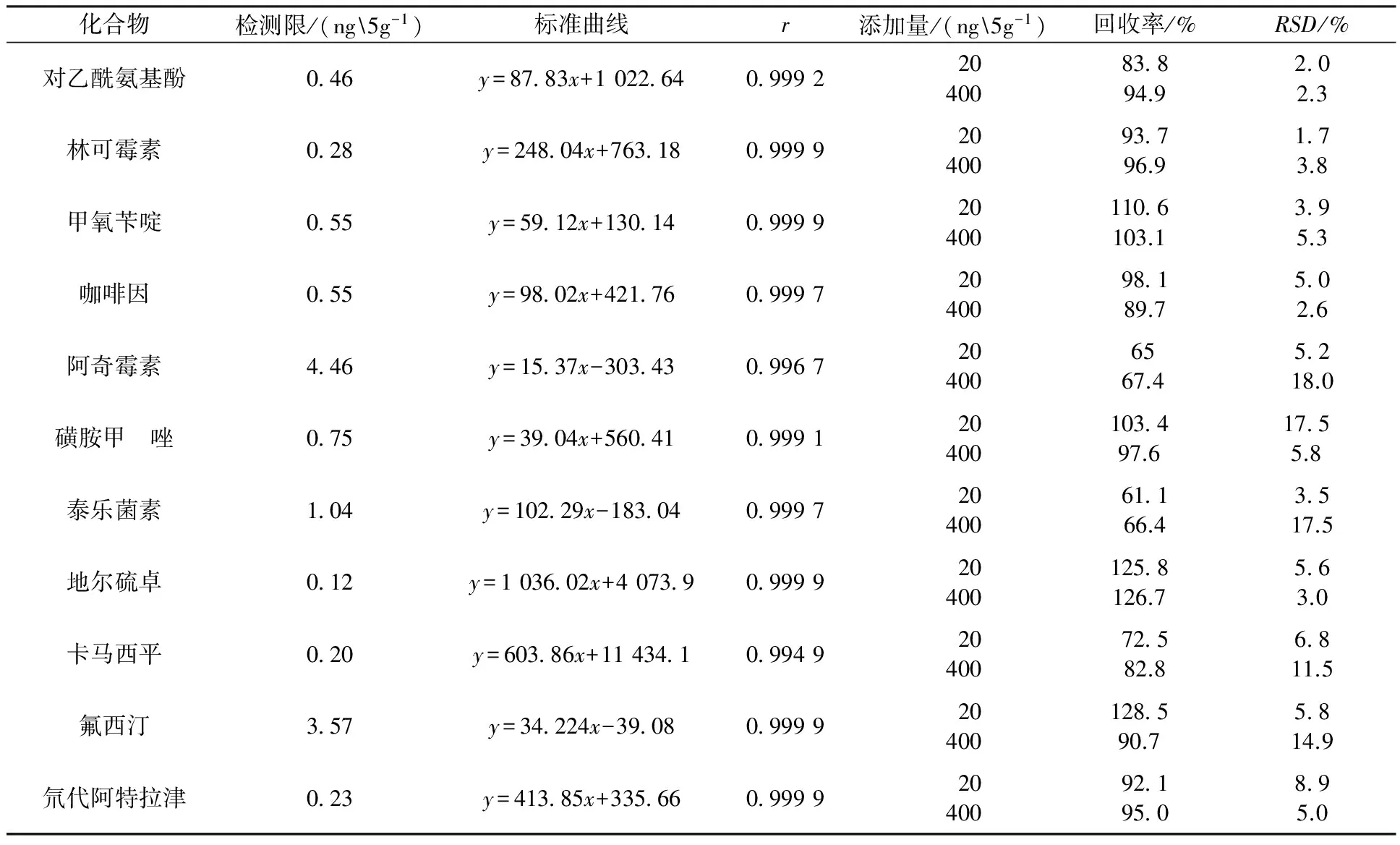
表3 10种PPCPs化合物及回收率指示物的检测限、标准曲线、回收率(n=5)及相对标准偏差

表4 10种PPCPs化合物徒骇河与独流减河表层沉积物中的干质量统计结果(几何均值) ng/g
3 结论
基于EPA 1694方法,对样品萃取与净化过程进行改进,采用超声波萃取与HPLC-MS/MS联用技术,建立了10种PPCPs同时分析的高灵敏度检测方法。该方法具有检测限低和回收率高的特点,并成功应用于海河流域表层沉积物的10种PPCPs污染物的检测。结果显示,咖啡因在独流减河中检测到的质量浓度为243.73 ng/g(干质量);对乙酰氨基酚、林可霉素、咖啡因和阿奇霉素在徒骇河中检测到的浓度分别为0.42、55.78、1.56、10.75 ng/g(均为干质量)。这些PPCPs的来源和污染现状有待进一步深入研究。
参考文献:
[1]Smital J, Wolf J, Sousa L L D. Estimation of genetic parameters of semen characteristics and reproductive traits in AI boars [J]. Animal Reproduction Science, 2005, 86(1): 119-130.
[2]周启星, 罗义, 王美娥. 抗生素的环境残留、生态毒性及抗性基因污染[J]. 生态毒理学报, 2007, 2(3): 243-251.
[3]J jemba P K. Excretion and ecotoxicity of pharmaceutical and personal care products in the environment [J]. Ecotoxicology and Environmental Safety, 2006, 63(1): 113-130.
[4]Hirsch R, Ternes T, Haberer K, et al. Occurrence of antibiotics in the aquatic environment [J]. Science of the Total Environment, 1999, 225(1/2): 109-118.
[5]Kolpin D W, Skopec M, Meyer M T, et al. Urban contribution of pharmaceuticals and other organic wastewater contaminants to streams during differing flow conditions [J]. Science of the Total Environment, 2004, 328(1): 119-130.
[6]Heberer T. Occurrence, fate, and removal of pharmaceutical residues in the aquatic environment: A review of recent research data [J]. Toxicology Letters, 2002, 131(1/2): 5-17.
[7]Stackelberg P E, Furlong E T, Meyer M T, et al. Persistence of pharmaceutical compounds and other organic wastewater contaminants in a conventional drinking-water-treatment plant [J]. Science of the Total Environment, 2004, 329(1): 99-113.
[8]Hamscher G, Pawelzick H T, Hper H, et al. Different behaviour of tetracyclines and sulfonamides in sandy soils after repeated fertilisation with liquid manure [J]. Environmental Toxicology and Chemistry, 2005, 24(4): 861-868.
[9]Kinney C A, Furlong E T, Kolpin D W, et al. Bioaccumulation of pharmaceuticals and other anthropogenic waste indicators in earthworms from agricultural soil amended with biosolid or swine manure [J]. Environmental Science & Technology, 2008, 42(6): 1 863-1 870.
[10]Agüera A, Fernández-Alba A R, Piedra L, et al. Evaluation of triclosan and biphenylol in marine sediments and urban wastewaters by pressurized liquid extraction and solid phase extraction followed by gas chromatography mass spectrometry and liquid chromatography mass spectrometry [J]. Analytica Chimica Acta, 2003, 480(2): 193-205.
[11]徐维海,林黎明,朱校斌. 水产品中14种磺胺类药物残留的HPLC法同时测定[J]. 分析测试学报,2004,23(5):122-124.
[12]Balizs G, Hewitt. Determination of veterinary drug residues by liquid chromatography and tandem mass spectrometry [J]. Analytica Chimica Acta, 2003, 492(1/2): 105-131.
[13]Cinquina A L, Roberti P, Giannetti F. Determination of enrofloxacin and its metabolite ciprofloxacin in goat milk by high-Performance liquid Chromatography with diode-array detection optimization and validation [J]. Journal of Chromatography A, 2003, 987(1/2): 221-226.
[14]徐冬梅,李青. 高效液相色谱法同时测定动物组织中3种抗生素残留量[J]. 中国公共卫生,2002,18(2):233-234.
[15]Dornlazabal V. Simultaneous extraction and determination of sulfadiazine and trimethoprim in medicated fish feed by High-Performance Liquid-Chromatography [J]. Journal of Chromatography A, 1993, 648(1): 183-186.
[16]US EPA 1694 PPCPs: Pharmaceuticals and personal care products in water, soil, sediment and bio-solids by HPLC/MS/MS [S].
[17]Marazuela M, Moreno-Bondi M. Multiresidue determination of fluoroquinolones in milk by column liquid chromatography with fluorescence and ultraviolet absorbance detection [J]. Journal of Chromatography A, 2004, 1034(1): 25-32.
[18]Johnston L, Mackay L, Croft M. Determination of quinolones and fluoroquinolones in fish tissue and seafood by high-performance liquid chromatography with electrospray ionisation tandem mass spectrometric detection [J]. Journal of Chromatography A, 2002, 982(1): 97-109.
[19]杨长志, 康庆贺, 马东升,等. 高效液相色谱法测定动物源性食品中甲氧苄氨嘧啶残留量[J]. 化学工程师,2005,(012):19-21.
[20]林海丹,谢守新,吴映璇. 高效液相色谱法同时测定鳗鱼及其制品中八种磺胺类药物[J]. 食品科学,2005,26(1):176-179.
[21]李孟玻, 张德云, 彭之见,等. 高效液相色谱法测定鱼肉中残留磺胺类药物[J]. 理化检验:化学分册,2006,42(8):611-619.
[22]Hospido A, Carballa M, Moreira M, et al. Environmental assessment of anaerobically digested sludge reuse in agriculture: Potential impacts of emerging micropollutants [J]. Water Research, 2010, 44(10): 3 225-3 233.
[23]Varga M, Dobor J, Helenkár A, et al. Investigation of acidic pharmaceuticals in river water and sediment by microwave-assisted extraction and gas chromatography-mass spectrometry [J]. Microchemical Journal, 2010, 95(2): 353-358.
[24]Williams M, Kookana R. Isotopic exchangeability as a measure of the available fraction of the human pharmaceutical carbamazepine in river sediment [J]. Science of the Total Environment, 2010, 408(17): 3 689-3 695.
