超声振荡辅助提取SPE-GC/MS法 测定袋泡茶中的49种农药残留
2014-03-22,,,
,,,
(1. 西安市疾病预防控制中心,陕西西安 710054;2. 陕西师范大学,化学化工学院,陕西西安 710062)
茶树在生长过程中难免受到病、虫侵害,因此经常需要使用各种农药,如果种植户不能正确使用农药,就可能造成已经收获的茶叶中存在着没有被降解的农药残留,这些残留的农药会随着食物链进入人体,给人体健康造成不利的影响。袋泡茶叶携带方便,消费量大,为了保护消费者健康,有必要对袋泡茶叶中的农药残留进行检测。农药残留前处理方法很多,为了提高提取和净化效果,科研工作者常常同时使用不同的前处理方法对农药进行处理。如超声辅助离子液体分散液液微萃取[1]、超声辅助萃取-分散液液微萃取[2]、微波辅助萃取-分散固相萃取法[3]、QuEchERS-凝胶渗透色谱[4]、超临界流体萃取-液相微萃取[5]、液液微萃取-多孔膜固相萃取法等[6],不同的前处理方法同时使用对农药的提取和净化效果更好。目前检测农药残留的仪器分析方法主要有气相色谱法[7]、气相色谱-质谱联用法(GC/MS)[8,9]、液相色谱法[10]、液相色谱-质谱联用法(LC/MS)[11]等。由于多数农药分子量小,沸点低,因此在农残检测中气质联用法更适用。气质联用法不但能够对复杂基质进行很好地分离,而且可以准确地定性定量,特别适合对复杂基质中的多种组份进行同时准确地测定。本研究采用超声振荡辅助提取-固相萃取-气质联用法对袋泡茶叶中的49种农药进行了检测方法研究,并对35份袋泡茶叶样品的农残进行了检测,初步了解了当前袋泡茶的农药残留情况,为今后政府监管和正确使用农药提供了实验数据依据。
1 材料与方法
1.1 材料与仪器
农药标准 农业部环境保护科研监测所;丙酮、正己烷 色谱纯,天津科密欧化学试剂有限公司。
7890A-5975C型气相色谱-质谱联用仪 美国安捷伦公司;SK-1型快速混合器、HY-4型振荡器 上海国华电器公司;TTL-DC II型氮吹仪 北京同泰公司;KH-700TDB型高频数控超声清洗器 昆山禾创超声仪器有限公司;固相萃取装置;固相萃取柱 CARB/NH2柱(500mg/500mg,6mL),迪马公司。袋泡茶样品 购于当地的市场和超市。
1.2 仪器条件
色谱条件:色谱柱:DB-1701(30m×0.25mm×0.25μm)毛细管柱;柱温:80℃,保持1.0min,以20.0℃/min的速度上升到220℃,保持1.0min,再以10.0℃/min的速度上升到270℃,保持23.0min;载气:氦气;恒流:1.0mL/min。不分流进样;进样口温度250℃。
质谱条件:离子源温度:230℃;接口温度:280℃;电子能量:70eV;扫描质量范围:50~500;阈值:150,溶剂延迟:4min。
1.3 农药前处理方法
1.3.1 农药的提取 准确称取经粉碎(至少20g)并全部过20目筛的样品5.0000g,放入100mL具塞三角瓶中,加入50.0mL丙酮,盖上塞子,先超声提取30min,再振荡提取30min,然后过滤,滤液通过装有5g无水硫酸钠的漏斗脱水后(采用无水硫酸钠脱水前,要先用少量的丙酮将无水硫酸钠浸湿,然后再对提取液脱水,最后再用少量丙酮冲洗无水硫酸钠),收集到100mL烧杯中,45℃以下低温氮吹近干,待过柱净化。
1.3.2 提取液的净化 以丙酮∶二氯甲烷=1∶1的混合溶液为淋洗液,将固相萃取(CARB/NH2/500mg/6mL)短柱置于SPE-24孔过滤装置上,先用5.0mL淋洗液淋洗柱子,再用3.0mL淋洗液采用超声和漩涡混合相结合的方法溶解提取物,将提取液过固相萃取柱,然后用3.0mL淋洗液冲洗烧杯,一并过柱,最后用3.0mL淋洗液洗脱萃取柱2次(采用固相萃取柱对样品净化时,小柱不能干涸而且洗脱液的流速不能太快,控制流速在1.5mL/min左右),收集于10.0mL比色管中,置于氮吹仪上45℃以下低温氮吹至近干,用丙酮定容至1.0mL,准确取1.0μL进样分析。
1.4 标准曲线的绘制
49种农药混混合标准用丙酮逐级稀释为0.125、0.25、0.5、1.0、1.5、2.0μg/mL的标准溶液,按确定的仪器条件以选择离子扫描(SIM)的方式采集色谱图,以浓度为横坐标、峰面积为纵坐标分别建立49种农药的标准曲线,并计算相关系数。
1.5 回收率实验
准确称取没有农药检出的绿茶样品5.0000g分别添加2.0μg/mL混合标准溶液250μL和1.5mL,即样品中农药的加标水平分别为0.1mg/kg和0.6mg/kg,平行测定4次进行回收率实验。
1.6 样品采集
本研究中的所有袋泡茶样品均从超市和市场购买。共采集了35份袋泡茶样品,其中红茶12份,绿茶9份,花茶5份,保健茶9份。
2 结果与讨论
2.1 农药的分离和定性
采用中等极性的DB-1701毛细管柱对49种农药组份进行分离,当色谱柱的初始温度大于80℃或者升温过快,有些农药,如γ-666和乙拌磷,抗蚜威和久效磷,马拉硫磷和杀螟硫磷,丙溴磷和杀扑硫磷就不能得到很好地分离。通过降低初始温度、调整程序升温速率和载气流速使上述较难分离的农药实现了分离,最终确定了1.2的仪器分析条件。根据1.2得到各农药的保留时间和特征离子(见表1)。49种农药混合标准物质(均为2.0μg/mL)的气相色谱-质谱选择离子色谱图见图1。
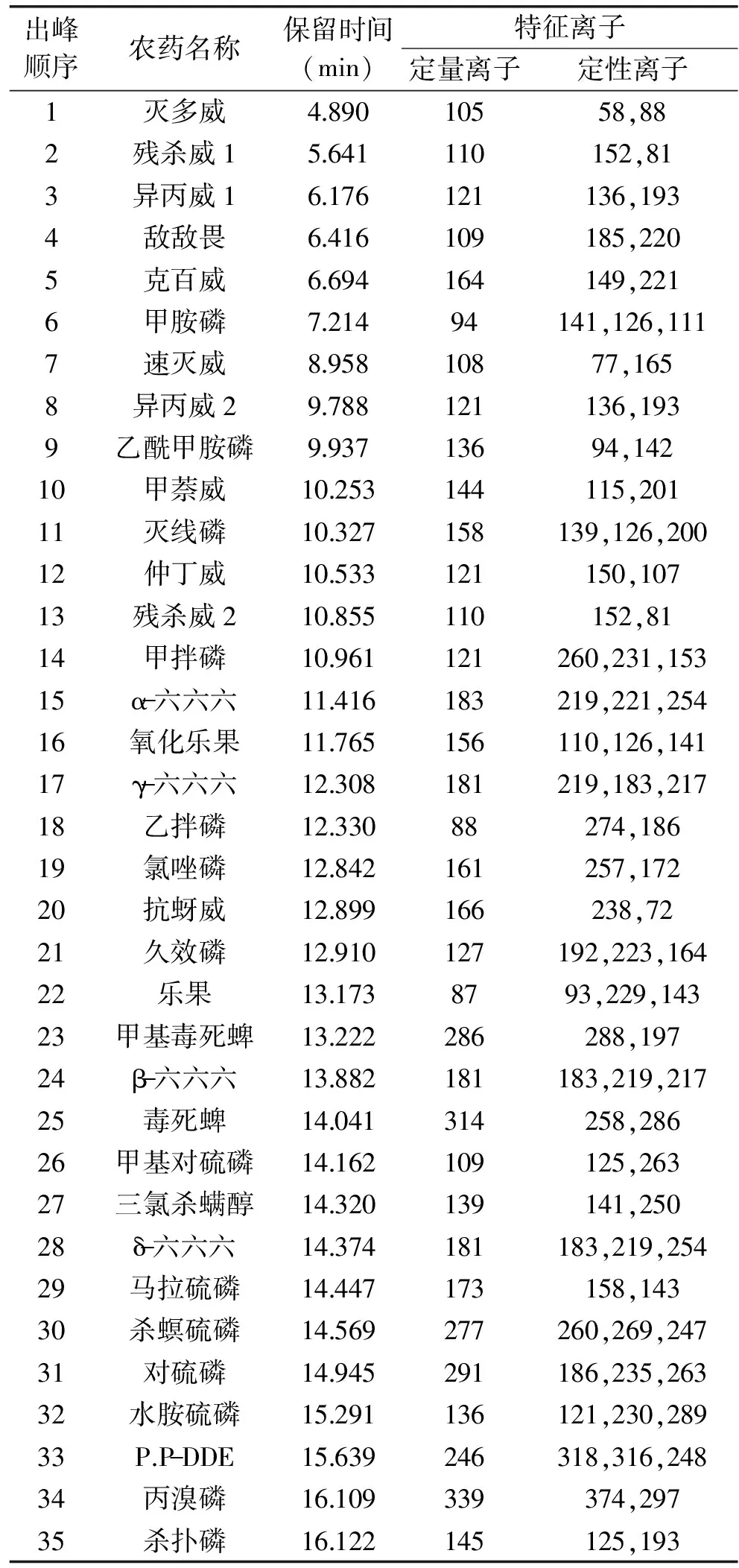
表1 49种农药的保留时间和特征离子Table 1 Retain time and characteristic ions of 49 pesticide residues
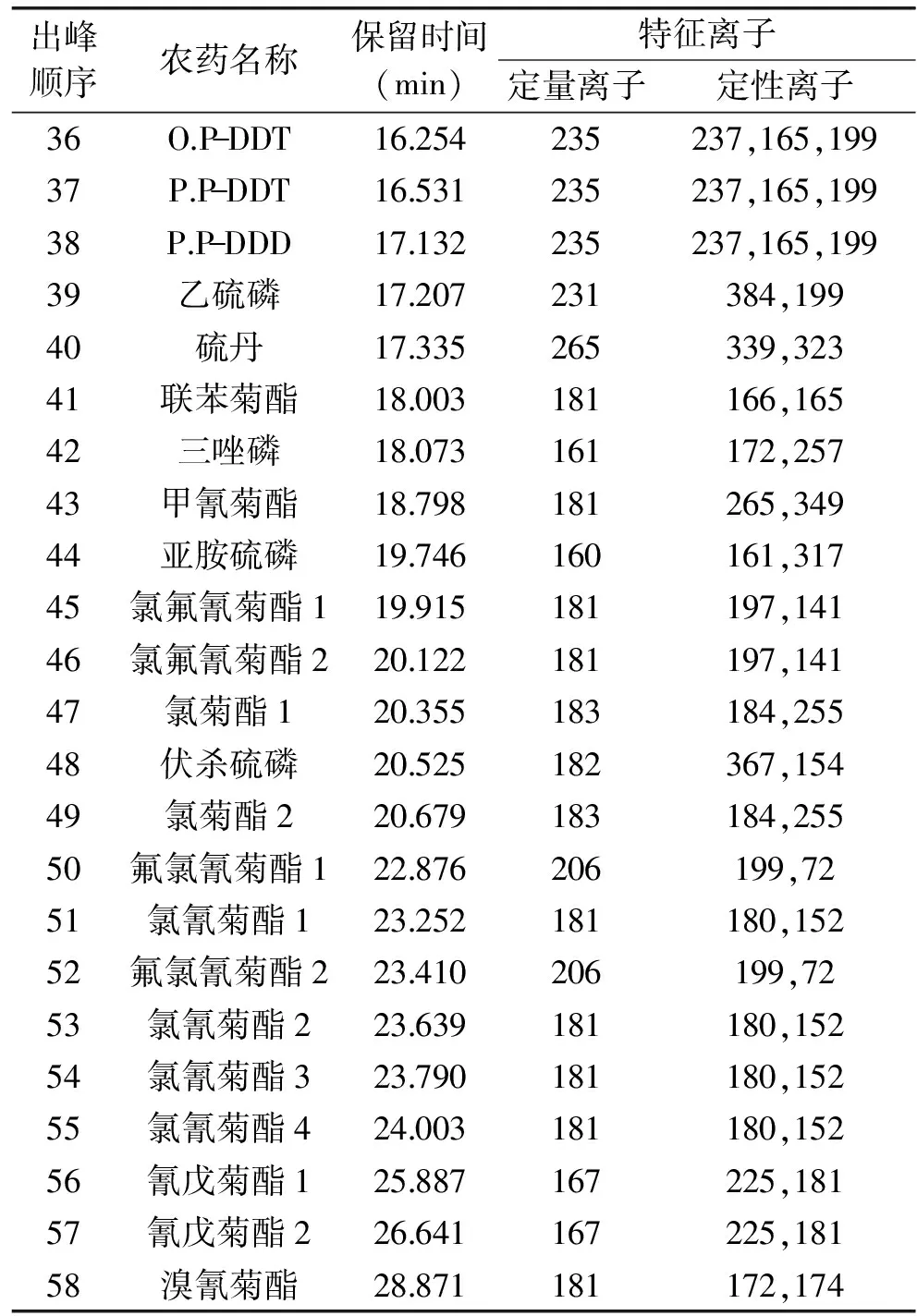
续表
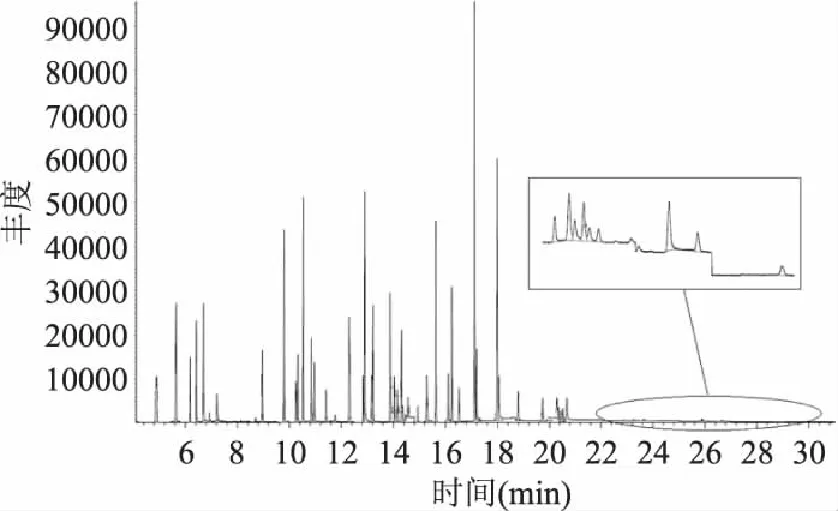
图1 49种农药混标的选择离子流图 Fig.1 Selective ion chromatogram of 49 pesticide residues
2.2 样品处理和分析细节的影响
2.2.1 提取液脱水分离 采用无水硫酸钠脱水时,应按1.3.1先用丙酮将无水硫酸钠浸湿,否则干的无水硫酸钠会吸附农药,造成回收率下降。
2.2.2 固相萃取的影响 0.4μg/mL的49种农药标准溶液分别以1.5mL/min和3.0mL/min按照1.3.2的方法进行洗脱实验发现,洗脱速度从1.5mL/min增加到3.0mL/min,49种农药的回收率下降到原来的63.8%~93.6%。对小柱淋洗后,如果小柱干涸后再上样,对茶叶样品的洗脱变得困难。
2.2.3 进样口清洁度的影响 样品分析时,进样口一旦被污染,甲胺磷、敌敌畏、氧化乐果、乙酰甲胺磷等极性农药的峰形变差。
2.3 线性方程、相关系数、检出限和回收率
本研究采用的SIM方法,大大降低了基线噪声,使其灵敏度显著提高。按照1.4得到各农药的线性方程和相关系数(0.9901~0.9998),以3倍信噪比计算各农药检出限在0.001~0.015mg/kg之间。通过添加0.1mg/kg和0.6mg/kg水平的回收实验发现:有机氯、拟除虫菊酯类农药和氨基甲酸酯类农药(除灭多威的回收有波动外)的回收率在75%~125%之间(拟除虫菊酯类农药的回收相对偏高),4次测定的相对标准偏差在2.8%~11.5%之间。但是,部分有机磷类农药的回收水平相对较差。其中,敌敌畏、氧化乐果、乙酰甲胺磷、甲胺磷、乐果回收率波动较大,有的甚至只有40%左右,4次测定的相对标准偏差在4.4%~32.8%之间。主要是因为这些有机磷类农药的性质不稳定,提取、净化和吹干过程对其有影响。具体结果见表2。
2.4 方法的准确性
本方法同时测定8种氨基甲酸酯类农药、10种有机氯类农药、8种拟除虫菊酯类农药和23种有机磷类农药。各类农药的性质差别较大,其中有机氯、拟除虫菊酯类农药和氨基甲酸酯类农药的稳定性较好,而部分有机磷类农药的稳定性较差。为了保证检测结果的准确性,每次测定样品时,均带加标样品及平行样,阳性样品均进行重复测定。上机检测时,10个样品之间插入一个标准,以便随时了解仪器的状态。
本方法采用的是气相色谱-质谱法,其可以通过保留时间、特征离子的质谱图与谱库中农药的标准质谱图比对2种方法对样品进行定性,因而定性的准确度相比气相色谱法有很大的提高。如图2是样品I(铁观音保健茶)和农药标准的比对色谱图,从图2可以看出,样品峰和联苯菊酯农药标准峰的保留时间均为17.991min,通过保留时间的一致性可以初步判断,样品I含有联苯菊酯,图3为样品的质谱图,与谱库中联苯菊酯的质谱图对比可知,样品峰的3个主要的特征离子为188、165、166,而且丰度比与谱库中一致,因此可以断定样品I中含有联苯菊酯,通过其与联苯菊酯农药标准色谱峰的面积比可以计算其含量。
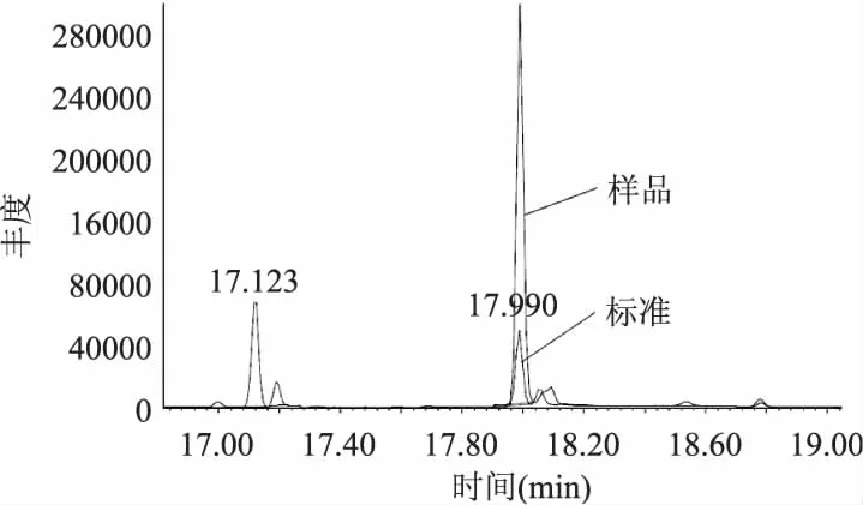
图2 样品I和标准的色谱图 Fig.2 Chromatograms of sample I and standard of pesticides
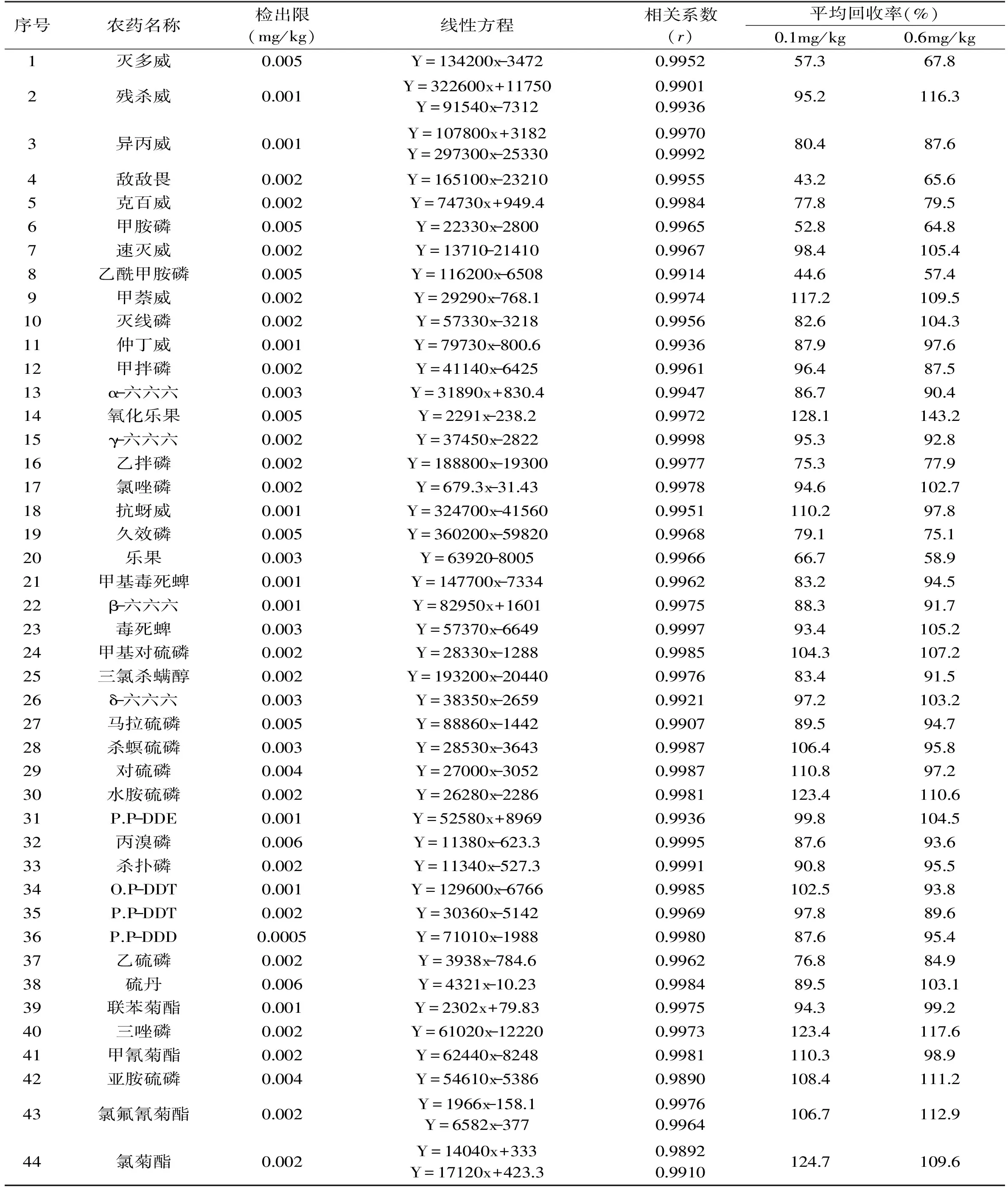
表2 各农药的线性方程、相关系数和回收率Table 2 The regression equations,correlation coefficients and recovery of pesticides
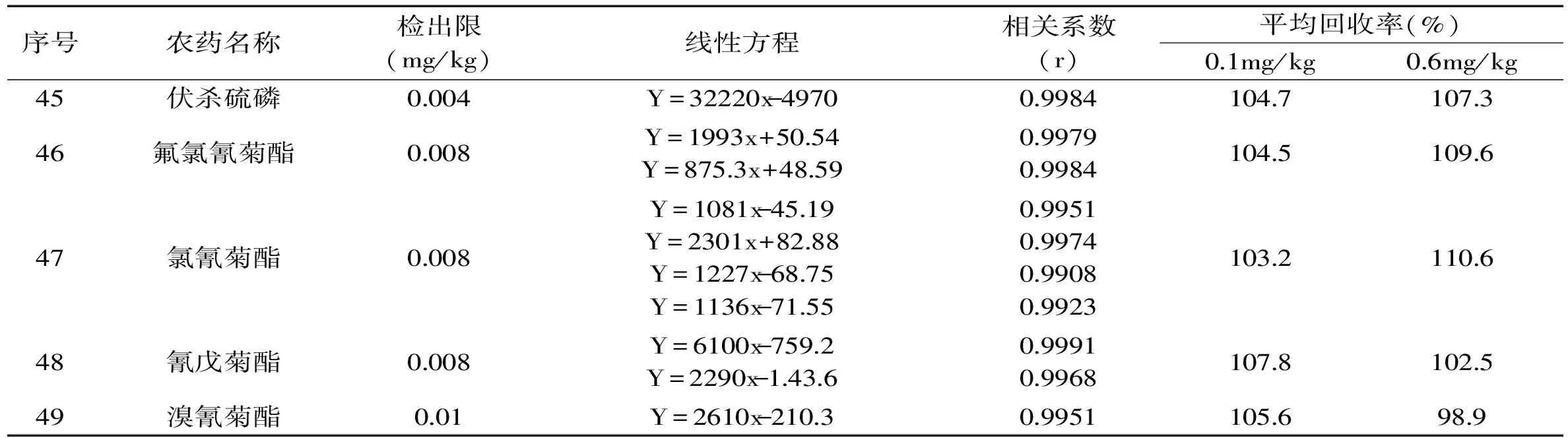
续表
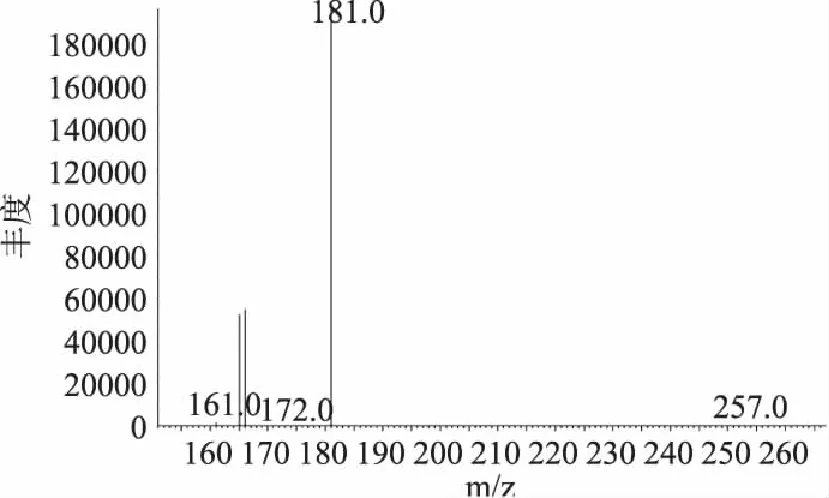
图3 样品I的质谱图 Fig.3 Mass spectrogram of sample I
但是,有时样品峰和农药标准峰的保留时间一致时,2个色谱峰也未必就代表同一物质。如图4是样品II(普洱保健茶)和农药标准的色谱图,从图4可以看出,样品峰和对硫磷农药标准峰的保留时间均为14.941min,如果仅采用气相色谱分析,通过保留时间定性,即可能判断样品II含有对硫磷。
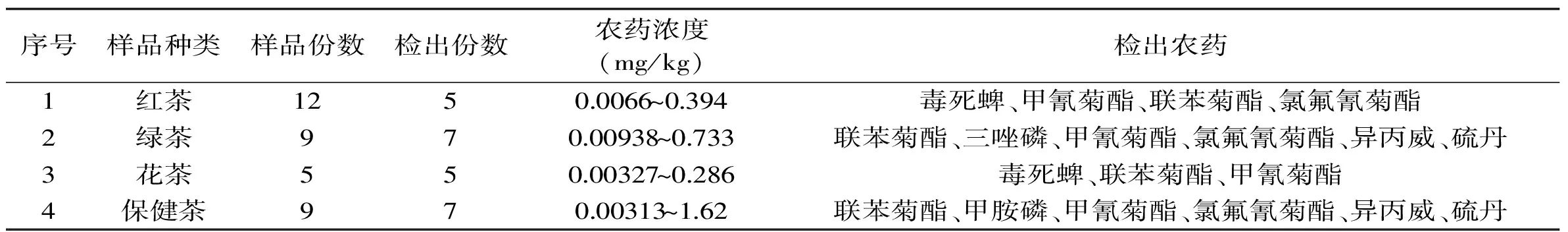
表3 袋泡茶的农药检出浓度和检出率Table 3 Concentration and positive rate of pesticide residues in tea bags
但是从样品II的质谱图(图5),可以看出它的4个主要的特征离子分别为235、186、263、291。4种特征离子235丰度最高,291丰度最低,即样品中的这个色谱峰主要是235离子的贡献,而谱库中对硫磷的色谱峰主要是291离子的贡献,因此,样品II中的这个色谱峰不能确认为对硫磷。
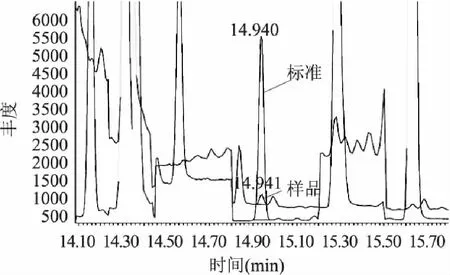
图4 样品II和标准的色谱图 Fig.4 Chromatograms of sample IIand standard of pesticides
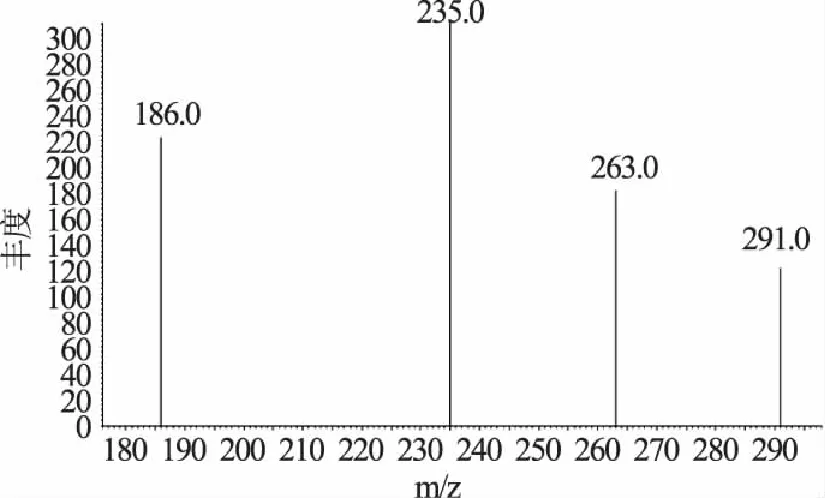
图5 样品II的质谱图 Fig.5 Mass spectrogram of sample II
从以上2个例子可以看出,单纯靠保留时间定性是不完全可靠的,很可能会做出假阳性的判断,而采用气相色谱-质谱法即可做出比较正确的判断。
2.5 样品测定
2.5.1 检测结果 通过对35份袋泡茶样品中的49种农药残留情况进行检测发现:红茶12份,阳性样品占41.67%;花茶共5份,全部有农药检出;绿茶和保健茶的阳性率均为77.78%,结果见表3。
受检样品中共检出8种农药,其中联苯菊酯、毒死蜱和甲氰菊酯检出率较高,8种农药的检出浓度和阳性样品见表4,各种农药的检出频次和检出百分比分别见图6和见图7。
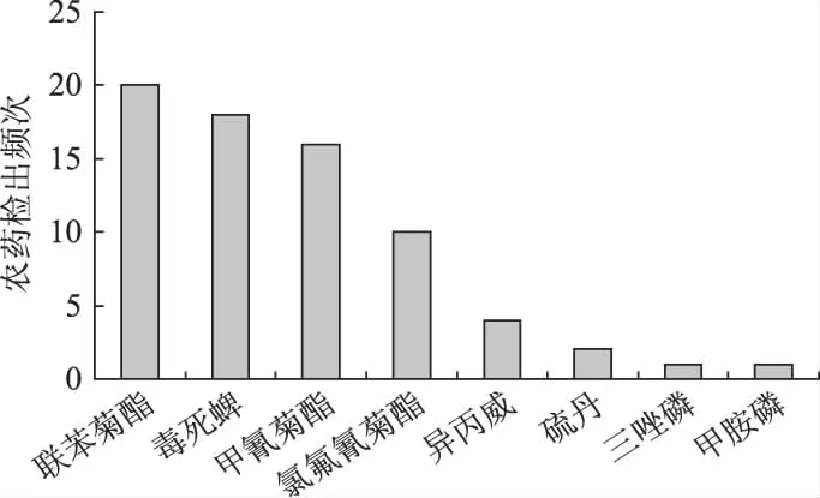
图6 农药检出频次 Fig.6 Frequencies of positive pesticide residues
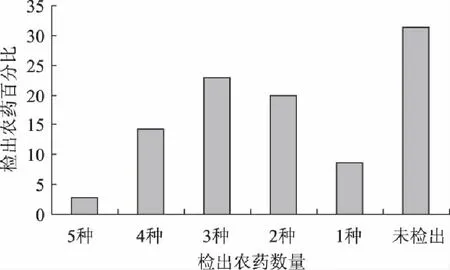
图7 茶叶样品农药检出百分比 Fig.7 The percentage of positive pesticides in tea bags
2.5.2 农药残留水平 GB2763-2012中规定甲氰菊酯、联苯菊酯和氯氟氰菊酯在茶叶中的限值分别为5、5、15mg/kg。而本研究中从袋泡茶样品中检出的所有农药的浓度均小于1.62mg/kg,因此,这3种农药在茶叶中的残留水平均未超标,并且检出浓度远小于标准的限值。
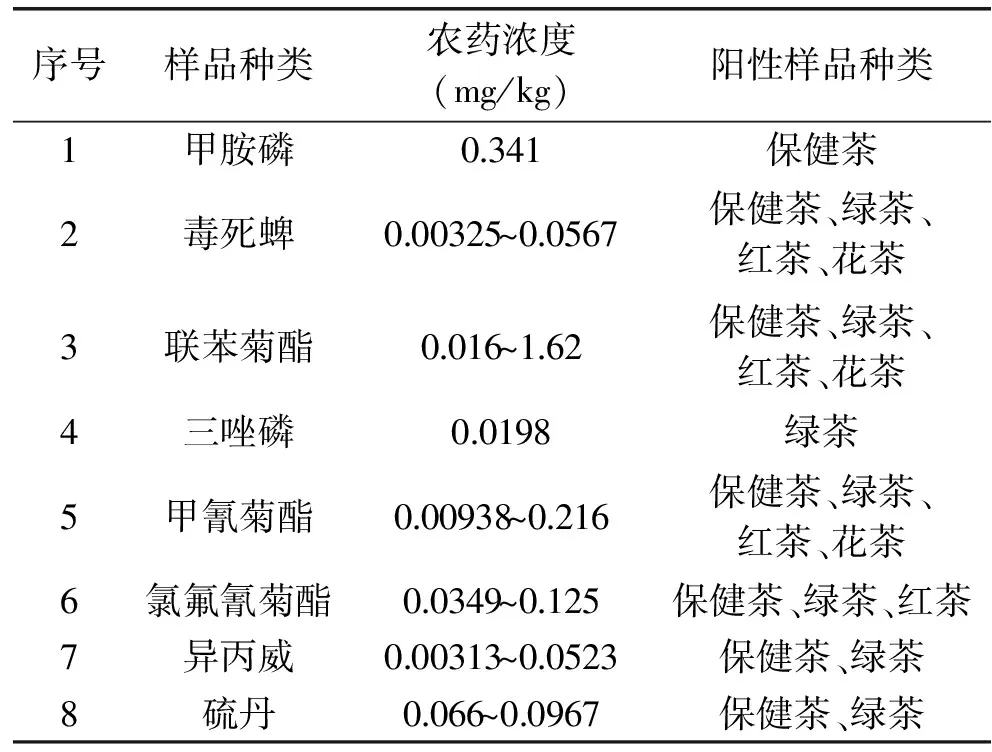
表4 样品中农药的检出浓度Table 4 Pesticides concentrations of positive tea bags
3 小结
本研究采用超声振荡辅助提取-固相萃取-气质联用法同时测定有机氯、拟除虫菊酯、有机磷和氨基甲酸酯类共4大类49种农药,采用超声振荡辅助提取-固相萃取方法对样品进行净化,建立的方法简单、快速,准确性好。通过对35份袋泡茶的检测发现,袋泡茶叶的农药检出率相对较高。根据GB2763-2012的限量规定,检出的甲氰菊酯、联苯菊酯和氯氟氰菊酯没有超标,浓度远低于限值规定。
[1]Zhang J H,Gao H X,Peng B,etal. Comparison of the performance of conventional,temperature-controlled,andultrasound-assisted ionic liquid dispersive liquid-liquid microextraction combined with high-performance liquid chromatography in analyzing pyrethroid pesticides in honey samples[J]. J chromatogr A,2011,1218:6621-6629.
[2]Bidari A,Ganjali M R,Norouzi P,etal. Sample preparation method for the analysis of some organophosphorus pesticides residues in tomato by ultrasound-assisted solvent extraction followed by dispersive liquid-liquid microextraction[J]. Food Chem,2011,126:1840-1844.
[3]Satpathya G,Tyagi Y K,Gupta R K. A novel optimised and validated method for analysis of multi-residues of pesticides in fruits and vegetables by microwave assisted extraction(MAE)-dispersive solid-phase extraction(d-SPE)-retention time locked(RTL)-gas chromatography-mass spectrometry with Deconvolution reporting software(DRS)[J]. Food Chem,2011,127:1300-1308.
[4]Lu D S,Qiu X L,Feng C,etal. Simultaneous determination of 45 pesticides in fruit and vegetable using an improved QuEChERS method and on-line gel permeation chromatography-gas chromatography/mass spectrometer[J]. J Chromatogr B,2012,895-896:17-24.
[5]Naeeni M H,Yamini Y,Rezaee M. Combination of supercritical fluid extraction with dispersive liquid-liquid microextraction for extraction of organophosphorus pesticides from soil and marine sediment samples[J]. J Supercrit Fluid,2011,57:219-226.
[6]Bedendo G C,Carasek E. Simultaneous liquid-liquid microextraction and polypropylene microporous membrane solid-phase extraction of organochlorine pesticides in water,tomato and strawberry samples[J].J Chromatogr A,2010,1217:7-13.
[7]Samadi S,Sereshti H,Assadi Y. Ultra-preconcentration and determination of thirteen organophosphorus pesticides in water samples using solid-phase extraction followed by dispersive liquid-liquid microextraction and gas chromatography with flame photometric detection[J]. J Chromatogr A,2012,1219:61-65.
[8]Wu J,Lu J,Wilson C,etal. Effective liquid-liquid extraction method for analysis of pyrethroid and phenylpyrazole pesticides in emulsion-prone surface water samples[J]. J Chromatogr A,2010,1217:6327-6333.
[9]Ma R,Rodríguez G,Otero R R,etal. Determination of 23 pesticide residues in leafy vegetables using gas chromatography-ion trap mass spectrometry and analyte protectants[J]. J Chromatogr A,2008,1196-1197:100-109.
[10]Boonchiangma S,Ngeontae W,Srijaranai S. Determination of six pyrethroid insecticides in fruit juice samples using dispersive liquid-liquid microextraction combined with high performance liquid chromatography[J]. Talanta,2012,88:209-215.
[11]M Arienzo,D Cataldo,L Ferrara. Pesticide residues in fresh cut vegetables from integrated pest management by ultra performance liquid chromatography coupled to tandem mass spectrometry[J]. Food Control,2013,31:108-115.
