UPLC-MS/MS检测原料奶中脱呋喃甲酰基头孢噻呋残留方法的建立
2014-02-25赵静,刘鹏,钱镭
赵 静,刘 鹏,钱 镭
(1.黑龙江东方学院食品与环境工程学部,黑龙江哈尔滨150086;2.国家乳业工程技术研究中心,黑龙江哈尔滨150086)
UPLC-MS/MS检测原料奶中脱呋喃甲酰基头孢噻呋残留方法的建立
赵 静1,2,刘 鹏2,*,钱 镭1,*
(1.黑龙江东方学院食品与环境工程学部,黑龙江哈尔滨150086;2.国家乳业工程技术研究中心,黑龙江哈尔滨150086)
目的:建立超高压液相色谱-串联质谱法(UPLC-MS/MS)测定原料奶中脱呋喃甲酰基头孢噻呋来间接衡量头孢噻呋残留的方法。方法:利用T3色谱柱,以含有0.02%的甲酸水-乙腈溶液为流动相进行梯度洗脱,采用二硫赤藓糖醇将原料奶中头孢噻呋分解为脱呋喃甲酰头孢噻呋(DFC),由碘乙酰胺衍生化为稳定的DFC乙酰胺形式(DCA),样品再经HLB固相萃取柱富集净化后,进行UPLC-MS/MS检测。结果:m/z 430.12>125.79为定量离子对,m/z 430.12>241.04为定性离子对,回收率在98.6%~102.2%,精密度(RSD)为2.51%~3.87%,最低检出限为0.45ng/mL。结论:该方法可用于原料奶中脱呋喃甲酰基头孢噻呋残留的测定。
超高压液相色谱-串联质谱法,脱呋喃甲酰基头孢噻呋,原料奶
头孢噻呋(ceftiofur),又名塞得福[1],属于β-内酰胺类抗生素[2],对革兰氏阳性菌、阴性菌甚至一些厌氧菌都具有很强的抗菌活性[3],因其抗菌谱广,抗菌活性强等特点,被美国和日本等国正式批准用于禽畜等的细菌性疾病的治疗[4],但其不规范的使用则会导致药物在动物机体内的残留,并由此而通过食物链进入人体,危害人体的健康。因此,为了保障人们的身体健康,许多国家都制定了严格的限量要求,欧盟2002年4月颁布了关于头孢噻呋在牛奶中的最大残留量(MRLS),是100μg/kg[5];我国农业部兽药使用规定,奶中的残留限量也是100μg/kg(农业部公告2002年第235号附录2)[6]。
牛奶中头孢噻呋残留量的检测方法主要有微生物法[7-8]、酶联免疫[9]、薄层色谱法[10-11]、高效液相色谱法[12-14],但是大部分检测方法都是直接检测头孢噻呋,而本研究是通过测定主要分解产物间接反映出头孢噻呋的量。20世纪90年后,随着检测技术的发展和检测要求的不断提高,液质联用法因其良好的适用性和高灵敏度已成为检测各种抗生素类药物的主要检测方法[15-22]。
头孢噻呋在动物体内很快代谢为脱呋喃甲酰基头孢噻呋(DFC),DFC较不稳定,因此本实验利用二硫赤藓糖醇将原料奶中头孢噻呋分解为脱呋喃甲酰基头孢噻呋(DFC),由碘乙酰胺衍生化为稳定的DFC
乙酰胺形式(DCA),样品再经HLB固相萃取柱富集净化后,采用超高压液相色谱-串联质谱法(UPLCMS/MS)法进行测定。该方法灵敏度高,净化效果好,重现性好,定量检出限为1.25ng/m L,低于头孢噻呋残留限量100ng/m L,可以满足对原料奶中头孢噻呋及其衍生物残留的检测要求。
1 材料与方法
1.1 材料与仪器
脱呋喃甲酰基头孢噻呋标准品(纯度99%)、二硫赤藓糖醇(纯度95%)、碘乙酰胺(纯度95%) TRC公司;乙腈、甲酸 色谱纯;硼酸、氯化钾、磷酸二氢钾等 均为分析纯;所用水 均为超纯净水。
2695高效液相色谱仪(包括四元泵、自动进样器、Empower色谱工作站,配2998二极管阵列检测器)、ACQUITY超高压液相色谱仪、Waters Xevo TQ质谱仪 美国Waters公司;QGC-12T型干热式氮吹仪 上海泉岛科贸有限公司;BS210S型电子天平 北京赛多利斯天平有限公司;超纯水仪 美国密理博公司;固相萃取装置 美国Mediwax公司。
1.2 溶液的配制
脱呋喃甲酰基头孢噻呋标准贮备液(100μg/m L):准确称取25mg去呋喃甲酰基头孢噻呋标准品,用乙腈-水=90∶10定容于25m L容量瓶中,于-20℃保存备用。
硼酸盐缓冲液(pH=9):准确称取四硼酸钠1.9000g,氯化钾0.3700g,加水约70m L后用盐酸调节pH至9.0,定容至100m L容量瓶中。
磷酸盐缓冲液(pH=7):准确称取0.3400g磷酸二氢钾,加水约70m L后用20%氢氧化钾调节pH至7.0,定容于100m L容量瓶中。
提取液:称取1.0g二硫赤鲜醇,用硼酸盐缓冲液溶解并稀释至250m L容量瓶中,现配现用。
碘乙酰胺溶液:称取5.0g碘乙酰胺,用磷酸盐缓冲液溶解并定容于50m L容量瓶中,现配现用。
25%磷酸溶液:取25m L磷酸,加水溶解并稀释至100m L。
1.3 实验方法
1.3.1 色谱条件 色谱柱:ACQUITY UPLC®HSST3 2.1mm×100mm,粒径1.8μm
流动相:A为0.02%甲酸/水,B为乙腈;流速:0.3m L/m in;梯度洗脱条件详见表1;柱温:30℃;样品温度:15℃;进样体积:10μL。

表1 UPLC-MS/MS梯度洗脱条件Table 1 The linear gradient condition of UPLC-MS/MS
1.3.2 质谱条件 电离方式:ESI(+);毛细管电压(Capillary voltages):3.0kV;锥孔电压:34V;源温度:150℃;去溶剂气温度(Desolvation Temperatures):450℃;去溶剂气流速(Desolvation gas flow):800L/hr;反吹气(Cone gas flow):50L/hr;碰撞气流速(Collision gas flow):0.3m L/m in;扫描模式:正离子扫描,多反映监测;脱呋喃甲酰基头孢噻呋母离子为m/z 430.12,子离子为m/z 125.79*和m/z 241.04。其中*为定量分析离子。
1.3.3 牛奶样品前处理 取5.0m L牛奶置于50m L离心管中,加入35m L提取液,混匀,于50℃恒温箱中保温15m in。加入10m L碘乙酰胺溶液,摇匀,室温下静置衍生30min。用25%的磷酸溶液调节pH至2.5~2.6,离心25m in(4℃,6000r/m in)。
依次用3m L甲醇、3m L水活化HLB萃取柱,取5m L上清液过柱,流速为1m L/m in,用3m L水淋洗小柱之后,用5m L 90%乙腈溶液洗脱,洗脱液用60℃氮吹吹干,残余物用1m L乙腈/水=90∶10溶液复溶,过0.22μm滤膜,供测定。
1.3.4 标准曲线的制备 将脱呋喃甲酰基头孢噻呋标准储备液,依次稀释为2、5、25、50、75、100、200ng/m L的标准工作液,衍生化处理后上机检测,每个标准溶液重复进样3次,以标准溶液浓度水平为横坐标,以待测物色谱图的平均峰面积为纵坐标,绘制标准曲线。
1.3.5 方法的专属性 分别取3个空白奶样,按样品处理方法项下操作,获得空白样品的色谱图,将浓度为10μg/m L的标准工作液加入空白基质中,依同法操作,获得相应的色谱图,将浓度1μg/m L的标准工作液依同法操作,获得相应的色谱图。
1.3.6 精密度和准确度的测定 分别在同一天和连续5d制备浓度为25、100、200ng/m L的头孢噻呋原料奶样品,每个浓度设5个重复,根据日内与日间标准偏差和相对标准偏差来评价方法的准确度和精密度。
1.3.7 回收率 配制标准浓度分别为25、100、200ng/m L的脱呋喃甲酰基头孢噻呋原料奶样品,每一浓度进行5个样本分析,获得相应的峰面积,以每一浓度处理的样品与理论添加标准品量的比值计算方法回收率。
1.3.8 灵敏度 向空白奶样中添加不同浓度的相应标准品溶液,以信噪比S/N=3时的最低浓度为最低定性检测限,S/N=10时的最低浓度为最低定量检测限。
2 结果与分析
2.1 色谱条件的优化
2.1.1 色谱柱的选择 根据脱呋喃甲酰基头孢噻呋的性质,选择了反相色谱柱,同时比较了常用的两种色谱柱:C18色谱柱和T3色谱柱,发现C18色谱柱杂质干扰峰较多,并且有严重的拖尾现象,而T3色谱柱可以很好的改善以上问题,使脱呋喃甲酰基头孢噻呋与其他杂质可以得到很好的分离,并且无拖尾现象,因此,本实验选择T3色谱柱作为分析柱。
2.1.2 流动相的选择 在UPLC-MS/MS分析中,反相髙效液相色谱常用的溶剂,如水、甲醇和乙腈十分有
利于电喷雾离子化。本实验比较了甲醇—水和乙腈—水为流动相时目标物的分离及响应情况,结果发现:乙腈—水作为流动相其分离效果更好,且其响应值较高。另外,由于脱呋喃甲酰基头孢噻呋在水溶液中容易形成游离态的离子,不利于反相色谱柱的保留和分离,所以在流动相中添加甲酸来改善色谱柱的保留和分离行为。本实验比较了水相中甲酸浓度为0.01%、0.02%、0.05%、0.1%、0.2%(v/v)对目标物离子化效率的影响,结果发现:当水相中含0.02%甲酸时,目标物的离子化效率最高,最终选择乙腈-水(含0.02%甲酸)溶液作为流动相。
2.1.3 流速的选择 根据UPLC-MS/MS分离原理,流动相流速过大和过小都会影响目标物与杂质的完全分离,同时会影响离子化效率和灵敏度,考虑分离效果、分离时间和灵敏度等综合因素,结合Esr源的推荐流速[23],设置流动相的流速为200μL/min。
2.1.4 柱温的选择 在合适的色谱柱和优化的流动相条件下,脱呋喃甲酰基头孢噻呋便可与杂质达到完全分离,因此本实验选择的柱温为30℃。
2.2 质谱条件的优化
用浓度为100ng/m L的单一标准品溶液,采用流动注射进样,在电喷雾正离子模式下进行质谱全扫描分析,在MSTune界面观察并调谐各离子源参数,优化电喷雾电压、载气流速、毛细管温度等参数,以丰度较大的离子峰m/z 430.12作为母离子,得到脱呋喃甲酰基头孢噻呋的母离子扫描图(图1);对母离子进一步进行子离子扫描,优化碰撞能量等参数,获得碎片离子峰,选择其中响应较强的两个子离子m/z 241.04和m/z 125.79作为监测该物质的碎片离子,并以峰形好、响应值低的一个离子m/z 125.79作为定量离子(图2)。优化的质谱条件下,得到脱呋喃甲酰基头孢噻呋的总离子流色谱图(图3)。

图1 脱呋喃甲酰基头孢噻呋的母离子扫描图Fig.1 Parent scan of desfuroylceftiofur

图2 脱呋喃甲酰基头孢噻呋的子离子扫描图Fig.2 Daughter scan of desfuroylceftiofur
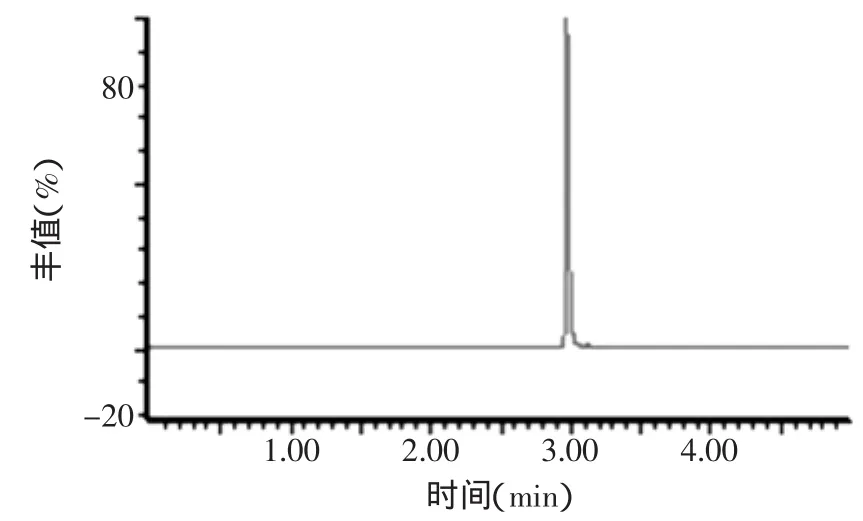
图3 脱呋喃甲酰基头孢噻呋的总离子流色谱图Fig.3 Total ions chromatogram of desfuroylceftiofur
2.3 脱呋喃甲酰基头孢噻呋标准曲线的制备
将脱呋喃甲酰基头孢噻呋储备液稀释以后,进行检测得出了脱呋喃甲酰基头孢噻呋的标准曲线,结果如图4所示,在脱呋喃甲酰基头孢噻呋为2~200ng/m L期间,标准曲线线性良好,相关系数为0.9991。
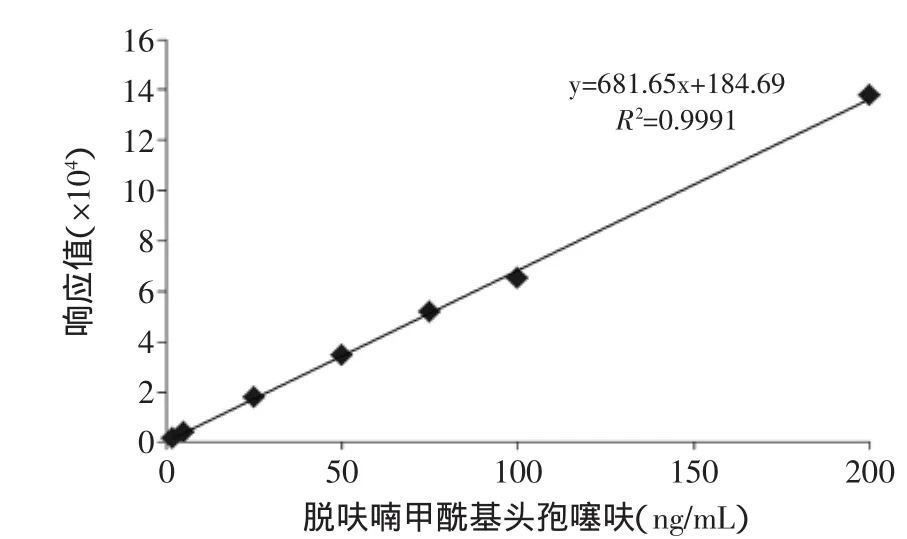
图4 脱呋喃甲酰基头孢噻呋的标准曲线Fig.4 Calibration curve of desfuroylceftiofur
2.4 方法的专属性
在上述分析条件下,空白、加标样品和标准品的色谱图见图5~图7,图5和图6对比可以发现空白样品中的物质对脱呋喃甲酰基头孢噻呋的检测没有干扰,从图7我们可以看出,脱呋喃甲酰基头孢噻呋的保留时间是3.0min。
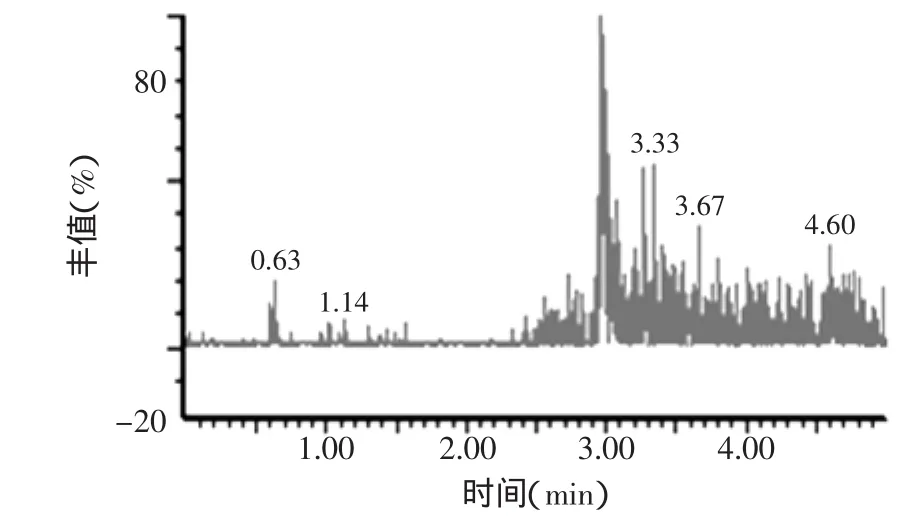
图5 空白奶样色谱图Fig.5 Chromatogram of blank milk samples
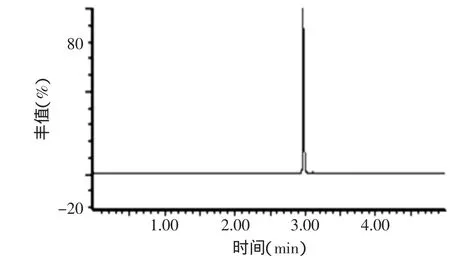
图6 空白奶样+头孢噻呋标准品色谱图Fig.6 Chromatogram ofblankmilk samplewith desfuroylceftiofur
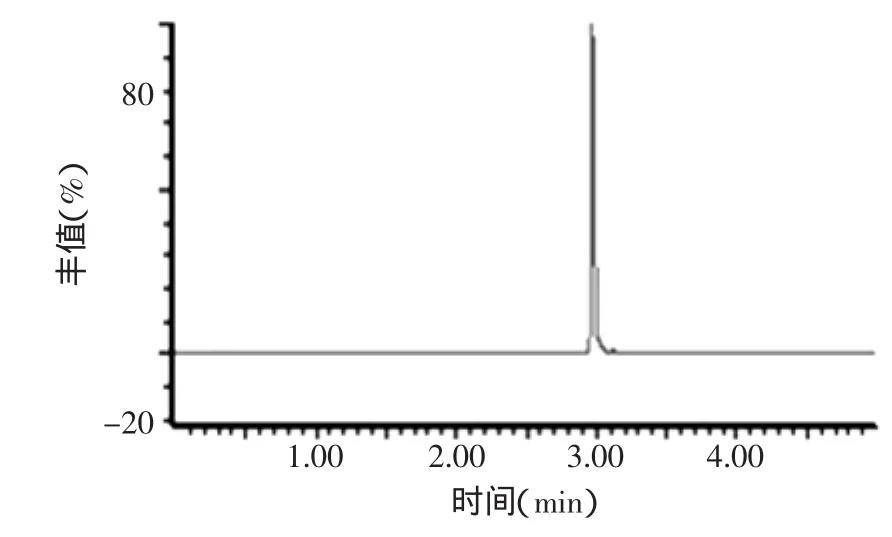
图7 头孢噻呋标准工作液色谱图Fig.7 Chromatogram of desfuroylceftiofur
2.5 精确度和准确度实验
分别选取了高、中、低三个浓度梯度脱呋喃甲酰基头孢噻呋,做日内和日间准确度和精密度实验,结果见表2。由表2可知,该方法准确度为2.5~3.4,精确度为2.51%~3.87%,准确度良好,精确度符合实验要求。

表2 脱呋喃甲酰基头孢噻呋在牛奶中的精密度和准确度Table 2 Determination of precision and accuracy ofdesfuroylceftiofur in blank milk
2.6 方法回收率实验
向空白牛奶中加入标准脱呋喃甲酰基头孢噻呋溶液,使脱呋喃甲酰基头孢噻呋浓度为25、100、200ng/m L,并上机测定,得出方法回收率见表3。
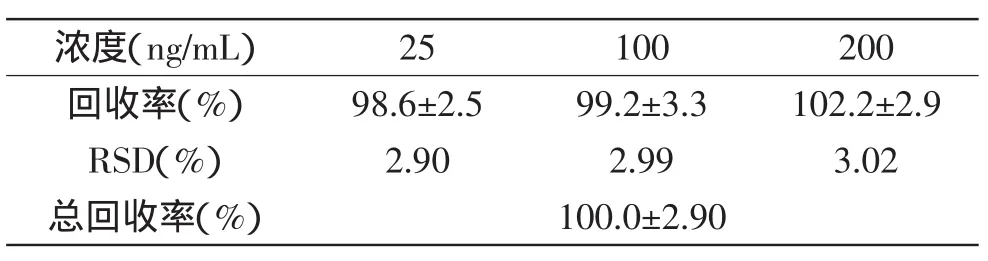
表3 牛奶中脱呋喃甲酰基头孢噻呋的方法回收率Table 3 Determination of recovery of desfuroylceftiofur in blank milk
2.7 灵敏度实验
根据信噪比(S/N)的值来确定最低定性检测限和最低定量检测限,结果表明该方法最低定性检出限为0.45ng/m L,定量检出限为1.25ng/m L。
3 结论
本文建立了脱呋喃甲酰基头孢噻呋在原料奶中残留检测的UPLC-MS/MS方法,采用梯度洗脱,色谱条件及质谱条件进行优化后,发现图谱基线平稳,脱呋喃甲酰基头孢噻呋的保留时间大约为3m in,且脱呋喃甲酰基头孢噻呋有较大的吸收峰,可以与原料奶中其他的杂质组分分离良好,该方法操作简便、灵敏度高,方法学考察验证了方法的准确性和可靠性,检测限为0.45ng/m L,可以满足目前国内外对原料奶中脱呋喃甲酰基头孢噻呋残留的检测要求。
[1]王慧,陈小军,刘伟,等.动物专用头孢噻呋[J].动物医学进展,2009,30(1):104-108.
[2]刘世娟.HPLC-MS/MS检测肌肉组织中多头孢菌素残留方法的建立[D].合肥:安徽农业大学,2012.
[3]昌莉丽,张春辉,肖传斌,等.RP-HPLC法测定头孢噻呋混悬注射液含量[J].西北农业学报,2010(6):44-47.
[4]王金涛.畜禽专用抗菌剂头孢噻呋[J].养殖技术顾问,2011(5):209.
[5]严凤,顾欣,李丹妮,等.奶牛乳房内灌注头孢噻呋钠的消除动力学研究[J].中国兽药杂志,2011,45(9):26-28.
[6]中华人名共和国农业部235号公告.《动物性食品中兽药最高残留限量》[S].北京:农业部,2002.
[7]Kress C,Seidler C,Kerp B,et al.Experiences with an identification and quantification program for inhibitor-positive milk samples[J].Analytica Chimica Acta,2007,586(1):275-279.
[8]于洁.兽用抗微生物药物残留质谱筛选方法研究[D].武汉:华中农业大学,2012.
[9]李铁柱,孙永海,郗伟东.受体分析结合酶联免疫检测牛乳中的头孢噻呋残留[J].高等学校化学学报,2008,29(3):473-476.
[10]Bilandži c'N,Kolanovi c'B S,Varenina I,et al.Veterinary drug residues determination in raw milk in Croatia[J].Food Control,2011,22(12):1941-1948.
[11]程瑶,商军,曹建东.紫外分光光度法快速测定盐酸头孢噻呋注射液的含量[J].上海畜牧兽医通讯,2011(5):10-11.
[12]Grzelak E M,Malinowska I,Choma IM.Determination of cefacetrile and cefuroxime residues in milk by thin-layer chromatography[J].Journal of Liquid Chromatography&Related Technologies®,2009,32(14):2043-2049.
[13]Nogueira D R,Silva LM,Arend M Z,et al.Development and validation of an HPLCmethod for determination of ceftiofur sodium[J].Chromatographia,2007,65(7-8):401-406.
[14]Zheng M M,Ruan G D,Feng Y Q.Hybrid organic-inorganic silicamonolithwith hydrophobic/strong cation-exchange functional groups as a sorbent formicro-solid phase extraction[J].Journal of Chromatography A,2009,1216(45):7739-7746.[15]Kantiani L,FarréM,BarcelóD.Analyticalmethodologies for the detection ofβ-lactam antibiotics in milk and feed samples[J].TrAC Trends in Analytical Chemistry,2009,28(6):729-744.
[16]Junza A,Amatya R,Barrón D,et al.Comparative study of the LC-MS/MSand UPLC-MS/MS for themulti-residue analysis of quinolones,penicillins and cephalosporins in cow milk,and validation according to the regulation 2002/657/EC[J].Journal of Chromatography B,2011,879(25):2601-2610.
[17]Martos P A,Jayasundara F,Dolbeer J,et al.Multiclass,Multiresidue Drug Analysis,Including Aminoglycosides,in Animal Tissue Using Liquid Chromatography Coupled to Tandem Mass Spectrometry†[J].Journal of Agricultural and Food Chemistry,2010,58(10):5932-5944.
[18]Turnipseed SB,Storey JM,Clark SB,et al.Analysis of Veterinary Drugs and Metabolites in Milk Using Quadrupole Time-of-Flight Liquid Chromatography-Mass Spectrometry[J]. Journal of Agricultural and Food Chemistry,2011,59(14):7569-7581.
[19]Kantiani L,FarréM,i Freixiedas JM G,etal.Determination of antibacterials in animal feed by pressurized liquid extraction followed by online purification and liquid chromatographyelectrospray tandem mass spectrometry[J].Analytical and Bioanalytical Chemistry,2010,398(3):1195-1205.
[20]Becker M,Zittlau E,Petz M.Quantitative determination of ceftiofur-related residues in bovine raw milk by LC-MS/MS with electrospray ionization[J].European Food Research and Technology,2003,217(5):449-456.
[21]Tang Y Y,Lu H F,Lin H Y,et al.Multiclass analysis of 23 veterinary drugs in milk by ultraperformance liquid chromatography-electrospray tandem mass spectrometry[J]. Journal of Chromatography B,2012,881:12-19.
[22]BeckerM,Zittlau E,PetzM.Residueanalysisof15 penicillins and cephalosporins in bovinemuscle,kidney and milk by liquid chromatography-tandem mass spectrometry[J].Analytica Chimica Acta,2004,520(1):19-32.
[23]TSQ Quantum液质联用仪使用手册[M].上海:赛默飞世尔科技(上海)有限公司,2007.
Establishment on UPLC-MS/MS detection methods fordesfuroylceftiofur in raw milk
ZHAO Jing1,2,LIU Peng2,*,QIAN Lei1,*
(1.Food and Environmental Engineering Department,Heilongjiang East College,Harbin 150086,China;2.National Research Center of Dairy Engineering and Technology,Harbin 150086,China)
Objective:A ultra-high pressure liquid chromatograph-tandem mass spectrometry (UPLC-MS/MS)was established for determination of desfuroylceftiofur residues in raw milk. Methods:T3 column was used witha mixed solution of 0.02% formic acid -acetonitrile as mobile phase. The cefliofur in bovine raw milk wasdecomposed by dithioerythritol to produce desfuroylceftiofur(DFC),which was further stabilized by derivatizationto desfuroylceftiofur acetamide(DCA) using iodoacetamide,then the samples were concentrated and purifiedby HLB SPE cartridge and determined by UPLC-MS/MS.Results:The quantitative ion was m/z 430.12>125.79.The qualitative ion was m/z 430.12>241.04. The recovery were 98.6%~102.2%,the relative standard deviations(RSD) were 2.51%~3.87%,the minimum detectable values were 0.45ng/mL. Conclusion:This method could beused for the routine analysis of desfuroylceftiofur residues in raw milk.
UPLC-MS/MS;desfuroylceftiofur;raw m ilk
TS201.1
:A
:1002-0306(2014)16-0092-05
10.13386/j.issn1002-0306.2014.16.011
2013-12-05 *通讯联系人
赵静(1989-),女,在读硕士研究生,研究方向:乳品质量与安全。
