大肠杆菌nmpC基因的快速敲除
2013-12-23张丹凤陈国平华秀庭陈阳
张丹凤 陈国平 华秀庭 陈阳
(1.漳州师范学院生物科学与技术系,漳州 363000;2.漳州市农业局种植业管理站,漳州 363000)
革兰氏阴性细菌的细胞壁有两层膜——外膜和内膜,外膜上有许多的外膜蛋白,其中包括外膜A蛋白、孔蛋白和脂蛋白等[1]。外膜蛋白处于最外层外膜上,它参与了多项生理功能,包括具有让外界物质进入的通道功能、帮助细菌摄取铁离子、参与革兰氏阴性细菌感染的粘附和炎症、激发宿主产生免疫保护性等生理功能,对于细菌适应外界环境和形成致病能力至关重要[2]。
NmpC蛋白质是一种外膜孔蛋白,组成磷脂双分子层中的微孔通道,可选择性地促进亲水分子通过革兰氏阴性细菌的外膜[3]。大肠杆菌 K-12△ompF和△ompC突变株中NmpC的表达量增加[4];进一步研究表明,大肠杆菌中NmpC蛋白质可以取代OmpF或者OmpC的功能,NmpC的表达会降低OmpF和OmpC孔蛋白的表达量[5]。同时大肠杆菌NmpC和鼠伤寒沙门氏菌(Salmonella typhimurium)中NmpC类似蛋白OmpD与OmpN蛋白功能极其相似[6]。NmpC还是一种新发现的与大肠杆菌耐药、耐热相关的外膜蛋白质[7,8]。除此之外,NmpC蛋白的表达受到TolC蛋白表达和nmpC启动子DNA成环的调控[9,10]。从蛋白质水平可以更充分地研究蛋白质的功能,但是目前还未见从基因删除的角度来进一步探讨NmpC蛋白质的功能。因此,基于Red重组系统和Xer重组系统构建了无标记的ΔnmpC基因删除菌株,旨在为深入探讨NmpC的功能及其分子机制奠定良好的基础。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 大肠杆菌 K-12 BW25113为本实室保存菌种;质粒pKD46(温度敏感型,含有阿拉伯糖启动子调控的gam、bet和exo基因,Ampr)和pSK-difGm(含有两边带有dif 位点[11]的庆大霉素抗性基因、Ampr和Gmr)均购于江苏锐阳生物科技有限公司。质粒pMD18-T simple vector 购于TaKaRa宝生物(大连)公司。
1.1.2 试剂和引物 Taq和Pfu DNA聚合酶、连接酶solution I、EcoR I和Hind III限制性内切酶、质粒提取、纯化试剂盒和胶回收试剂盒为均为 TaKaRa宝生物(大连)公司产品。抗生素庆大霉素和氨苄青霉素购于上海生工生物工程股份有限公司。大肠杆菌基因删除试剂盒购于江苏锐阳生物科技有限公司。本研究所用引物序列如表1所示。

表1 PCR扩增所用引物
1.2 方法
1.2.1 PCR扩增nmpC片段 提取大肠杆菌 K-12 BW25113基因组,利用引物nmpC1和nmpC2进行PCR扩 增,PCR体 系 为:基 因 组DNA 2 μL、2.5 mmol/L dNTPs 4 μL、2.5 mmol/L Mg2+2 μL、25 mmol/L nmpC1和nmpC2各0.3 μL、Taq DNA聚合酶0.4 μL、10×缓冲液 5 μL和ddH2O 36 μL;扩增程序为:95℃ 5 min;94℃ 30 s,56℃ 1 min,72℃ 75 s,共30个循环;72℃ 10 min。取PCR产物进行电泳和胶回收。
1.2.2 nmpC基因删除用线性化DNA片段的制备 将胶回收的nmpC片段,通过连接酶solution I与pMD18-T simple vector进行连接,转化大肠杆菌DH5α,提取质粒,经Hind III单酶切验证,获得重组质粒pMD-nmpC。该重组质粒用EcoR I酶切,切除nmpC片段中150 bp片段,经电泳胶回收目的片段pMD-nmpC1。进一步通过Pfu DNA聚合酶补平pMD-nmpC1酶切切口,乙醇沉淀回收目的片段。补平后的目的片段用碱性磷酸酶CIAP进行去磷酸化得到pMD-nmpC2,防止平末端发生自身连接。Sma I酶切pSK-difGm,获得difGm片段。difGm片段与pMD-nmpC2连接,构建重组质粒T-nmpC∷difGm。经Hind III单酶切验证,获得含有突变盒的重组质粒T-nmpC∷difGm。
1.2.3 突变盒片段的PCR 扩增 将含有突变盒片段的重组质粒T-nmpC∷difGm用Hind III酶切线性化,电泳回收线性的重组质粒片段,去除环形质粒。以该片段为模板,利用引物nmpC1和nmpC2 PCR扩增获得突变盒nmpC∷difGm,胶回收PCR 产物获得高浓度突变盒片段。
1.2.4 电转化突变盒片段入大肠杆菌细胞 通过CaCl2转化法将pKD46转化大肠杆菌 K-12 BW25113。将含有pKD46的大肠杆菌 K-12 BW25113接种于LB平板,30℃培养24 h。挑取单菌落接种于50 mL LB液体培养基,30℃条件下200 r/min过夜培养。取0.5 mL 培养液接种于50 mL含有氨苄青霉素(100 μg/mL)和2 mmol/L L-阿拉伯糖的新鲜LB 液体培养基,30℃、200 r/min培养至A600值约为0.7。将培养液迅速置于冰上冷却20 min,4℃、4 000 r/min离心10 min 收集菌体。倾倒培养基,并轻柔地将菌体重新悬浮于20 mL预冷的10%(V/V)甘油溶液中,离心收集菌体,并将菌体重悬于20 mL预冷的10%(V/V)甘油溶液中,并重复该步骤2次,将上清液倾倒,并将菌体用残留的约100 μL 10%(V/V)甘油溶液重悬。轻柔地将50 μL菌体溶液与35 μL纯化后的突变盒PCR 产物混合,置于0.1 cm预冷的电转杯中,电转仪1.8 kV进行电击,电击时间为4-5 ms。迅速加入1 mL 含有1 mmol/L L-阿拉伯糖的LB 液体培养基,将菌液在37℃培养1 h后,涂布于含有庆大霉素(30 g/mL)的LB 固体培养基上筛选重组菌株。
1.2.5 阳性转化子的验证 挑取庆大霉素抗性筛选获得的转化子,利用引物Gm1和nmpC3进行菌落PCR验证大肠杆菌 K-12 BW25113和重组菌株,获得阳性转化子。
1.2.6 Xer重组酶系介导的庆大霉素抗性基因的去除 将阳性转化子接种于LB 液体培养基,37℃培养12 h,然后转接于新鲜LB液体培养基继续培养12 h。稀释培养液至适当浓度,涂布LB平板,37℃培养。挑取单菌落同时点种于含庆大霉素的固体LB平板和不含庆大霉素的普通固体LB平板,挑选庆大霉素抗性丢失的转化子(在dif 位点进行了Xer 重组的细胞失去了庆大霉素抗性),并用nmpC1和nmpC2引物进行进一步的菌落 PCR 扩增验证,最终获得没有抗生素标记的基因删除菌株。
2 结果
2.1 试验方案设计
经Vector NTI软件分析nmpC基因内部有2个EcoR I位点,2个位点中间有150 bp的碱基序列,通过酶切去除这150 bp碱基,并经Pfu补平后与经Sma I酶切获得的difGm片段进行连接。在2个EcoR I位点外围各扩增500 bp碱基作为同源臂用于整合,在Red 重组酶的作用下进行双交换,将目的基因替换,进一步在自身Xer重组酶的介导下,进行2个dif位点的重组将抗生素标记基因去除。
2.2 重组质粒pMD-nmpC的构建
利用引物nmpC1和nmpC2对nmpC进行PCR扩增,获得与预期1 016 bp的片段大小相当的目的片段,结果如图1-A所示。将该目的片段与pMD18-T simple vector进行连接,获得pMD-nmpC重组质粒,重组质粒图谱如图2-A所示。提取pMDnmpC重组质粒,经Hind III酶切,图1-B第2泳道显示酶切片段大小约为3 703 bp,成功获得了含有nmpC基因片段的重组质粒。
2.3 重组质粒T-nmpC∷difGm的构建
大量提取pMD-nmpC,利用EcoR I酶切去除nmpC基因中150 bp的片段,得到约3.6 kb的pMD18-nmpC1片段(图1-B第3泳道)。Sma I酶切pSK-difGm,获得difGm片段,difGm片段与去磷酸化的、补平酶切切口的pMD18-nmpC2连接,获得重组质粒T-nmpC∷difGm,其物理图谱如图2-C所示。用Hind III对T-nmpC∷difGm进行单酶切,如图3-A所示获得大小为4 585 bp的目的条带,与预期大小一致。

图1 nmpC基因PCR扩增片段(A)及pMD-nmpC重组质粒(B)
2.4 nmpC∷difGm的PCR 扩增
用Hind III对T-nmpC∷difGm重组质粒进行酶切,进行胶回收,获得线性化的T-nmpC∷difGm。以此目的片段为模板,通过引物nmpC1和nmpC2进行PCR扩增获得突变盒nmpC∷difGm,获得大小约1 900 bp的目的条带,与预计的片段大小相当(图3-B)。
2.5 ΔnmpC基因删除菌株的鉴定
胶回收突变盒nmpC∷difGm,获得高浓度的纯化的突变盒,将其电转化入大肠杆菌 K-12 BW25113细胞中。庆大霉素抗性筛选获得的转化子,通过引物Gm1和nmpC3经菌落PCR验证确认,获得片段大小约1.2 kb的目的片段(图4第1泳道)。依次传代去除庆大霉素抗性和pKD46,获得不具有庆大霉素抗性的目标菌株。经菌落PCR验证(引物nmpC1和nmpC2)只获得大小约850 bp的片段(图4第5泳道)。结果显示,成功获得了ΔnmpC基因突变无标记菌株。
3 讨论
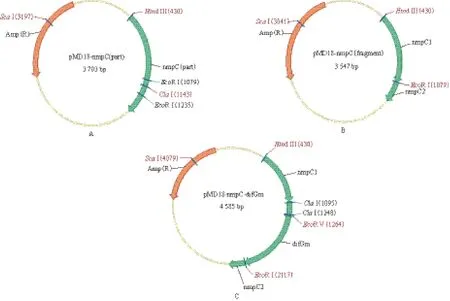
图2 重组质粒物理图谱

图3 T-nmpC∷difGm酶切(A)及nmpC∷difGm PCR扩增(B)产物

图4 nmpC基因删除转化子PCR鉴定电泳图谱
基因删除技术是一种从分子水平上定向改变生物活体遗传信息的试验手段,对于研究基因和蛋白功能等生命科学的重大问题十分重要。基于λ噬菌体Red重组酶作用的同源重组技术逐渐成为基因工程研究的热点之一,BaBa等[12]利用这种方法,已经对大肠杆菌 K-12中3 985个目的基因进行删除,并由此建立起新的大肠杆菌基因库(http://ecoli.aist-nara.ac.jp/)。但是该方法在去除抗生素标志时,需要进行带有相应重组酶基因的外源质粒的转化,使试验周期延长。Bloor 等[11]采用Red重组系统-Xer重组系统,实现靶基因重组后抗生素标记高效删除,方法简便。目前该技术已经在多种微生物细胞成功获得无抗生素标记的基因删除菌株,包括大肠杆菌、幽门螺杆菌(Helicobacter pylori)、分枝杆菌(Mycobacteria)和枯草芽孢杆菌(Bacillus subtilis)等[13,14]。本研究运用该技术利用大肠杆菌细胞中自身Red重组系统-Xer重组系统,通过PCR验证获得无抗生素标记的基因删除菌株ΔnmpC。NmpC的表达量与OmpF和OmpC的表达呈负相关[4,5],期望利用该基因删除菌株来深入探讨NmpC的缺失表达对OmpF和OmpC表达的影响,甚至与调控OmpF和OmpC表达的EnvZ/OmpR双调节系统之间的关系。NmpC是新发现的与大肠杆菌适应逆境的外膜蛋白[7,8],通过ΔnmpC可以进一步验证它在大肠杆菌耐受不利环境过程中的作用,及与其它相关功能蛋白间错综复杂的网络关系,均将促进NmpC外膜蛋白功能的阐明。
4 结论
首次成功地在大肠杆菌Red 重组系统和Xer重组系统的介导下,获得大肠杆菌ΔnmpC无抗生素标记基因删除菌株,该基因删除菌株将有利于进一步探讨NmpC在大肠杆菌不同生理过程的作用及其它功能。
[1] Solov’eva TF, Novikova OD, Portnyagina OY. Biogenesis of β-Barrel integral proteins of bacterial outer membrane [J]. Biochemistry (Mosc), 2012, 77(11):1221-1236.
[2] Gimeno C, Navarro D, Savall F, et al. Relationship between outer membrane protein profiles and resistance to ceftazidime, imipenem, and ciprofloxacin in Pseudomonas aeruginosa isolates from bacteremic patients [J]. European Journal of Clinical Microbiology and Infectious Diseases, 1996, 15(1):82-85.
[3] Hindahl MS, Crockford GW, Hancock RE. Outer membrane protein NmpC of Escherichia coli:pore-forming properties in black lipid bilayers [J]. Journal of Bacteriology, 1984, 159(3):1053-1055.
[4] Lee DR, Schnaitman CA, Pugsley AP. Chemical heterogeneity of major outer membrane pore proteins of Escherichia coli [J]. Journal of Bacteriology, 1979, 138(3):861-870.
[5] Prilipov A, Phale PS, Koebnik R, et al. Identification and characterization of two quiescent porin genes, nmpC and ompN, in Escherichia coli BE [J]. Journal of Bacteriology, 1998, 180(13):3388-3392.
[6] Singh SP, Miller S, Williams YU, et al. Immunochemical structure of the OmpD porin from Salmonella typhimurium [J]. Microbiology, 1996, 142(11):3201-3210.
[7] Gautam A, Vinson HM, Gibbs PS, et al. Proteomic analysis of multidrug resistant Escherichia coli strains from scouring calves [J]. Veterinary Microbiology, 2011, 151(3-4):363-371.
[8] Ruan L, Pleitner A, Gänzle MG, et al. Solute transport proteins and the outer membrane protein NmpC contribute to heat resistance of Escherichia coli AW1.7 [J]. Applied and Environmental Microbiology, 2011, 77(9):2961-2967.
[9] Morona R, Reeves P. The tolC locus of Escherichia coli affects the expression of three major outer membrane proteins [J]. Journal of Bacteriology, 1982, 150(3):1016-1023.
[10] Coll JL, Heyde M, Portalier R. Expression of the nmpC gene of Escherichia coli K-12 is modulated by external pH. Identification of cis-acting regulatory sequences involved in this regulation [J]. Molecular Microbiology, 1994, 12(1):83-93.
[11] Bloor AE, Cranenburgh RM. An efficient method of selectable marker gene excision by Xer recombination for gene replacement in bacterial chromosomes [J]. Applied and Environmental Microbiology, 2006, 72(4):2520-2525.
[12] Baba T, Ara T, Hasegawa M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants:the Keio collection [J]. Molecular Systems Biology, 2006, 2:2006.0008.
[13] Debowski AW, Gauntlett JC, Li H, et al. Xer-cise in Helicobacter pylori:one-step transformation for the construction of markerless gene deletions [J]. Helicobacteria, 2012, 17(6):435-443.
[14] Cascioferro A, Boldrin F, Serafini A, et al. Xer site-specific recombination, an efficient tool to introduce unmarked deletions into mycobacteria [J]. Applied and Environmental Microbiology, 2010, 76(15):5312-5316.
