红色荧光蛋白mCherry中插入外源短肽位点的研究
2013-12-23梁俊婷李鹿之陈少鹏焦浈
梁俊婷 李鹿之 陈少鹏 焦浈
(1.郑州大学 离子束生物工程省重点实验室,郑州 450052;2.中国科学院合肥物质科学研究院技术生物与农业工程研究所,合肥 230031)
自1962年Shimomura等[1]首次从维多利亚多管水母(Aequoria victoria)中分离出绿色荧光蛋白(avGFP)后,到目前为止,通过一系列体外分子进化的手段发展出几乎覆盖整个荧光光谱的GFP突变体,如蓝色(Blue)、青色(Cyan)、黄色(Yellow)荧光蛋白[2-5]。绿色荧光蛋白主要作为报告基因应用于生命科学领域。然而,1997年Abedi等[6]将20个氨基酸残基的短肽插入到GFP松散的Loop位点处,其中大部分插入的位点都严重影响荧光蛋白的折叠和发光,但同时筛选出了少数合适的插入位点,在插入外源短肽后仍能正常发射荧光。这种体外进化GFP的方式不同于以往改造GFP的方式,传统的方法主要采用一轮或多轮的点突变改造GFP荧光蛋白。随后,Baird等[7]对增强型绿色荧光蛋白(EGFP)的每一个位点做了更为系统和精确的筛选,其可插入外源短肽的位点基本上与GFP保持一致,但是其他的荧光蛋白或者GFP突变体是否也具有类似的或不同的耐受外源短肽插入的位点呢?
1999年,红色荧光蛋白drFP583[8](商品名是DsRed,558 nm/583 nm)首次被发现,极大丰富了荧光蛋白的光谱多样性。较之GFP及其突变体,红色荧光蛋白drFP583的激发和发射波长大大增加,而且具有在组织成像方面背景荧光低等诸多优势。因此,红色荧光蛋白引起了研究人员的广泛关注。但是野生型的drFP583体内外均易形成四聚体且成熟速度慢[9],这些缺陷极大限制了它在生物学上的应用。为了克服这些缺点,研究人员随后通过多轮的随机突变和定点突变改造,在2002年首次得到了红色荧光蛋白drFP583的单体mRFP1[10]。与野生型drFP583相比,mRFP1具有更长的激发和发射波长(584 nm/607 nm),其荧光亮度更强,成熟速度更快。在mRFP1的基础上,Shaner等[11]通过多轮随机和定点突变得到了mCherry。优化后的mCherry比mRFP1荧光亮度更亮,成熟时间更短,激发和发射波长更长(587 nm/610 nm)。因其诸多优良性能,mCherry备受研究人员的青睐。
已有研究表明mCherry类似GFP,可以插入外源短肽[12]。为了获得更多的更合适的插入位点,本研究使用mCherry为靶荧光蛋白,旨在通过转座子的突变系统寻找mCherry中可供外源短肽插入的位点。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒和培养基 大肠杆菌感受态细胞Trans I及质粒pUC19均购自北京全式金生物公司。LB培养基、SOC培养基和LB培养基配方参考分子克隆实验指南(第3版)。LB/Agar/Amp培养基(Amp终浓度是200 μg/mL),LB/Agar/Amp/Kan培养基(Amp终浓度是200 μg/mL,Kan终浓度为10 μg/mL)。
1.1.2 主要的试剂 Not I 、EcoR I 和Sal I限制性酶以及DNA Ladder Marker购买于TaKaRa公司。高保真酶KOD Plus 购自ToYoBo公司。普通Taq酶购自北京TIANGEN生物公司。IPTG(异丙基-β-D-硫代吡喃半乳糖苷)贮存浓度为100 mmol/L,工作浓度为1 mmol/L。T4连接酶购自NEB公司。PCR产物纯化试剂盒、质粒提取试剂盒、胶回收试剂盒均为Axygen公司产品。寡核苷酸引物合成和DNA测序由上海生工生物公司完成。转座子随机突变试剂盒Mutation Generation SystemTMKit(F-701)购自Finnzymes公司。
1.2 方法
1.2.1 质粒构建 用于扩增mCherry的上下游引物分 别 为:F:5'-TCAGACGAATTCGGTACCGGCGGC TCCATGGTGAGCAAGGGCGAGGAGG-3',R:5'-AG ATTGTCGACGGTACCGCCCTTGTACAGCTCGTCCATG CCGC-3'。分别在上游引物加上限制酶EcoR I,下游引物加上限制酶Sal I,mCherry经PCR扩增后,经EcoR I 和Sal I 酶切。回收mCherry酶切片段连接到经EcoR I 和Sal I 酶切的质粒pUC19上,得到重组质粒pUC-MC。转化入大肠杆菌感受态细胞Trans I中,使用质粒提取试剂盒提取质粒pUC-MC。
1.2.2 以转座子试剂盒为基础的突变体系统 按照转座子随机突变试剂盒Mutation Generation SystemTMKit说明书,使用具有Kan抗性的MuA转座子进行体外转座反应。将自身DNA随机转座到已经构建好的质粒pUC-MC(靶DNA)上。体外转座反应体系按照说明书要求,在1.5 mL的离心管中分别加入1 μL转座子,120 ng目的DNA(质粒pUC-MC),4 μL Buffer,1 μL MuA转座酶,最后加入超纯水至终体积20 μL。轻轻混匀后30℃反应1 h,75℃水浴10 min,灭活MuA转座酶。
1.2.3 mCherry 及其突变体的获得 取2 μL转座酶体外转座反应系统的反应混合物,加入到50 μL Trans I感受态细胞中,转化后涂布在LB/Agar/Amp培养板上,37℃培养过夜。收集所有的细菌克隆后提取质粒。EcoR I 和Sal I 酶切后,琼脂糖凝胶电泳,产生4条明亮且大小不同的条带(图1-A)。这4条带大小分别为3.8 kb(2.7 kb质粒+1.1 kb转座子),2.7 kb(空质粒),1.8 kb(0.7 kb mCherry +转座子)和0.7 kb(mCherry)。回收1.8 kb和2.7 kb的条带,连接后转化,涂板,37℃培养过夜后,收取所有克隆,最终得到在mCherry中插入转座子的细菌库,提质粒,得到pUC-MC/TS。由于转座子序列两端均含有特定的Not I酶切位点,质粒pUC-MC/TS 经Not I酶切后,转座子基因基本被切除,仅留下包括Not I酶切位点在内的15 个碱基,得到质粒pUC-MC/15 bp。
1.2.4 mCherry及其突变体的诱导表达 将构建好的质粒系统pUC-MC/TS和pUC-MC/15 bp分别转入大肠杆菌感受态细胞Trans I,在含有200 μg/mL Amp或(和)10 μg/mL Kan的TB培养液中,37℃,180 r/min条件下培养12 h。然后将菌液1∶50接种在含有相应抗生素的TB培养液中,在37℃,180 r/min下培养菌液直到OD600达到0.4。加入IPTG诱导使其终浓度为1 mmol/L,在16℃,180 r/min条件下诱导mCherry荧光蛋白,8 h后流式细胞仪检测其荧光变化。
1.2.5 流式细胞仪检测及分选 将诱导后表达mCherry的菌液12 000 r/min离心去上清,用磷酸盐缓冲液(PBS)洗2遍。调整菌液OD600值为0.2后,用AriaIII流式细胞仪(BD)进行分选和检测(激发波长:561 nm,发射波长:610 nm)。3次平行试验后,根据其相对荧光强度,观察突变体相对于野生型mCherry的荧光变化。
1.2.6 mCherry突变体测序 在流式细胞仪分选阳性菌液培养后挑取单克隆菌斑,测序。各测序结果与mCherry序列比对后确认有15个碱基插入到mCherry序列即可认定为阳性克隆。
1.2.7 统计分析 试验数据来自至少3次重复试验,结果的表示方法为:x-±s。数据组之间的显著性分析用 students’t test方法,P<0.05为显著性差异。
2 结果
2.1 mCherry短肽插入突变体库的构建
利用转座子随机突变系统将转座子(Kan抗性)随机插入到质粒pUC-MC,使用mCherry两端特定的限制酶EcoR I和Sal I 酶切后得到4条不同大小的DNA片段(图1-A),将含有转座子序列的mCherry片段(1.8 kb)连接到pUC19片段(2.7 kb)上得到突变体库pUC-MC/TS(图1-B)。在此突变体库的基础上,使用限制酶Not I切除突变体pUC-MC/TS上的转座子序列,在原位置残留15个碱基。这样便得到15个碱基仅插入到mCherry序列的质粒突变体库pUC-MC/15 bp(图1-C)。

图1 突变体库pUC-MC/TS和pUC-MC/15 bp的构建
2.2 mCherry短肽突变体的筛选
适当的条件下诱导表达mCherry短肽突变体后,使用流式细胞分选仪(ArialIII,BD)分选出阳性细菌(图2),表达红色荧光蛋白的大肠杆菌如图2-B P3所示。扩大培养后,即获得插入短肽后仍发荧光的突变体群。
2.3 mCherry短肽突变体的获得
将流式细胞仪分选出的阳性菌培养后涂板,挑取单克隆后测序。测序的各突变体序列与原始mCherry序列比对后,确定15个碱基插入到mCherry序列及其插入位置,然后分析其在荧光蛋白mCherry二级结构中的插入位置。
测序结果显示,有15个碱基插入到mCherry序列的克隆共有53个,占所有测序的单克隆总数(85个)的64%,即筛选mCherry短肽突变体的阳性率约64%。同时,共有13个mCherry短肽突变体诱导后仍发射红色荧光,其插入位置分别位于mCherry氨基酸序列的第K9、E10、F11、F65、G90、G116、K121、E148、M150、L157、G171、V187和T223氨基酸残基位置处,分别简称为m9、m10、m11、m65、m90、m116、m121、m148、m150、m157、m171、m187和m223(图3)。
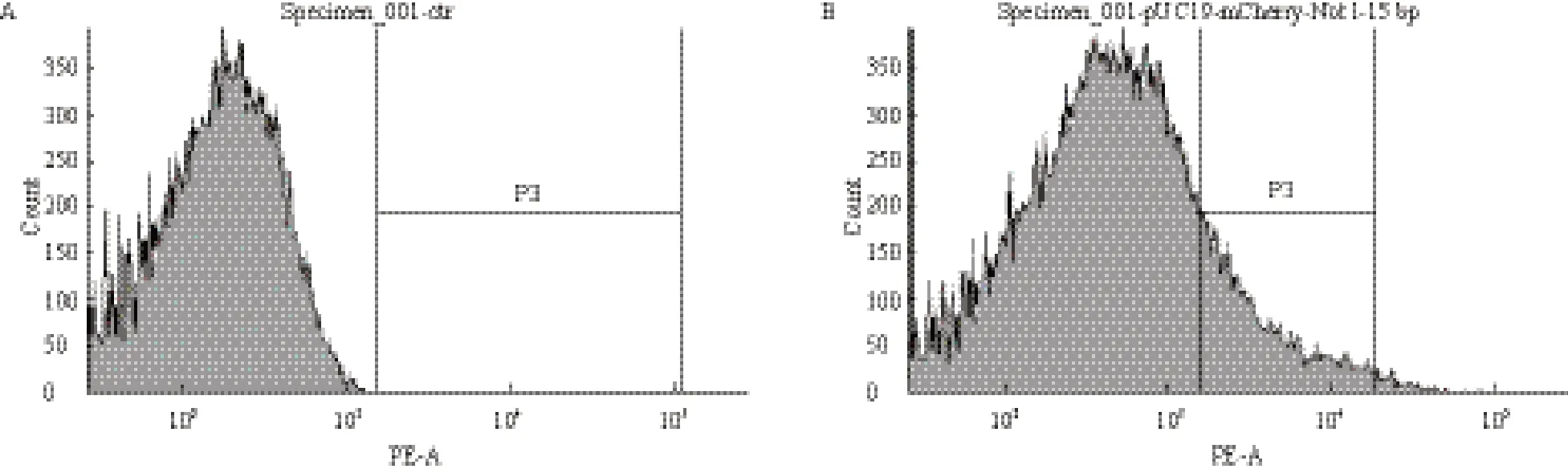
图2 流式细胞仪筛选诱导后表达质粒突变体pUC-MC/15bp的大肠杆菌
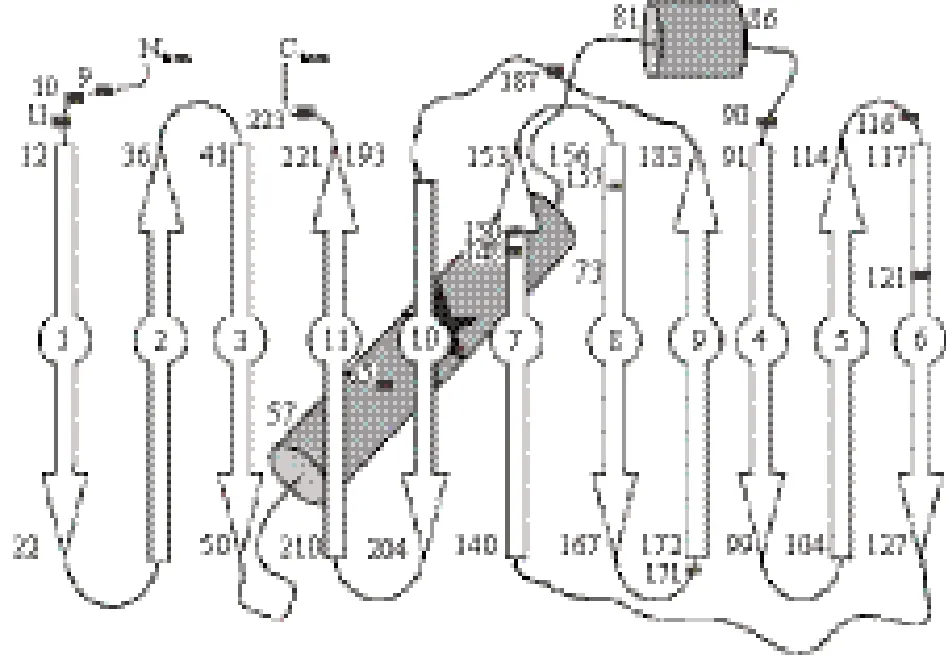
图3 5个外源氨基酸所在mCherry二级结构中相应氨基酸残基的位置(方框所示)
2.4 mCherry短肽突变体荧光亮度的测定
将上述13个mCherry短肽突变体及野生型mCherry在相同诱导条件下诱导后,流式细胞仪检测它们在大肠杆菌内的表达。mCherry短肽突变体的荧光亮度与野生型mCherry(未插入的荧光强度为100%)相比明显减弱(图4)。
3 讨论

图4 质粒突变体pUC-MC/15bp中15个碱基插入mCherry序列的氨基酸位数对应mCherry突变体的相对荧光强度
Mu噬菌体是目前研究DNA转座的理想模型之一,其转座机理[13]已有详尽报道。本研究采用MuA转座酶催化的转座子突变系统,将转座子随机插入到荧光蛋白mCherry中,经转座子两端特定的Not I 限制酶切后,剩余15个碱基残留在mCherry序列中。但是,15个碱基插入到mCherry序列后,荧光蛋白mCherry的荧光大部分都已经消失,只有在若干个特定位点处mCherry仍保留较强荧光。这些特定的位点主要分布在N/C末端,第3、4个β折叠之间,第5、6个β折叠之间,第8、9个β折叠之间,第9、10个β折叠之间以及第6、7、8个β折叠上。Li等[12]证明了mCherry可耐受外源短肽插入的位点主要分布在第1、2个β折叠之间,第6、7个β折叠之间,第9、10个β折叠之间,但认为最耐受插入外源肽段的位点是在第9、10个β折叠之间的Loop结构。本研究显示,虽然在第9、10个β折叠之间的Loop结构(第187位氨基酸残基)处的突变体m187仍能发出56%的相对荧光,但并无充分的证据证实此Loop结构最耐受外源肽段的插入。第9、10个β折叠之间的Loop中第V187位氨基酸残基处插入的m187突变体的荧光强度与插入其他Loop结构处的突变体短肽相比荧光最亮;第5、6个β折叠之间的荧光蛋白突变体亮度最低,约为野生型mCherry荧光亮度的15%。因此本研究证明此Loop结构中耐受外源短肽插入的程度较弱。
另外,本研究在β折叠上也筛选出4个不同的插入位点,与插入到松散的Loop结构中的各个位点相比,荧光蛋白突变体的亮度总体降低。但在第7个β折叠上有两个位点处(第E148和M150位)的荧光蛋白突变体均能发出较亮的荧光。这说明与其他β折叠相比,这两个位点处或其附近的位点耐受度较强。迄今为止,仍未有相关的文献报道,系统筛选出荧光蛋白mCherry的β折叠上所有耐受外源短肽插入的位点。本研究使用突变转座系统,首次将β折叠柱上可插入位点系统的筛选出来,共有4个可插入位点。同时本研究还发现,位于荧光蛋白mCherry的发色体(MYG)附近的第F65位突变体m65,荧光强度约为原来的52%。
本研究证明的可插入位点不仅包括已有相关报道的附近位点,如V187处与Li等[12]报道的K184位都在第9、10个β折叠柱之间的Loop结构,而且证实了更多不同的位点,这极大地增加了荧光蛋白mCherry中可插入的位点的数目,为试验的进一步设计(如荧光双分子互补技术)提供更多可选择的位点。同时,也为改造荧光蛋白作为生物感受器或者插入更长外源短肽提供了更多可参考的位点。
4 结论
采用转座子随机突变系统最终将5个氨基酸短肽随机到荧光蛋白mCherry中,共筛选出13个特定插入位点。这些插入外源短肽的mCherry突变体仍然能发出较强的荧光,其中第9、10个β折叠柱间的Loop结构耐受外源短肽插入的能力较强;相反,在第5、6个β折叠柱间耐受度较弱。
[1] Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea [J]. Journal of Cellular and Comparative Physiology, 1962, 59(3):223-239.
[2] Tsien RY. The green fluorescent protein [J]. Annual Review of Biochemistry, 1998, 67:509-544.
[3] Patterson G, Day RN, Piston D. Fluorescent protein spectra [J]. Journal of Cell Science, 2001, 114(5):837-838.
[4] Rizzo MA, Springer GH, Granada B, et al. An improved cyan fluorescent protein variant useful for FRET [J]. Nature Biotechnology, 2004, 22(4):445-449.
[5] Griesbeck O, Baird GS, Campbell RE, et al. Reducing the environmental sensitivity of yellow fluorescent protein. mechanism and applications[J]. Journal of Biological Chemistry, 2001, 276(31):29188-29194.
[6] Abedi MR, Caponigro G, Kamb A. Green fluorescent protein as a scaffold for intracellular presentation of peptides [J]. Nucleic Acids Research, 1998, 26(2):623-630.
[7] Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins [J]. Proc Natl Acad Sci, 1999, 96(20):11241-11246.
[8] Matz MV, Fradkov AF, Labas YA, et al. Fluorescent proteins from nonbioluminescent Anthozoa species [J]. Nature Biotechnology, 1999, 17(10):969-973.
[9] Baird GS, Zacharias DA, Tsien RY. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral [J]. Proc Natl Acad Sci, 2000, 97(22):11984-11989.
[10] Campbell RE, Tour O, Palmer AE, et al. A monomeric red fluorescent protein [J]. Proc Natl Acad Sci, 2002, 99(12):7877-7882.
[11] Shaner NC, Campbell RE, Steinbach PA, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein [J]. Nature Biotechnology, 2004, 22(12):1567-1572.
[12] Li Yankun, Sierra AM, Ai H, et al. Identification of sites within a monomeric red fluorescent protein that tolerate peptide insertion and testing of corresponding circular permutations [J]. Photochemistry and Photobiology, 2008, 84(1):111-119.
[13] Haniford DB, Chaconas G. Mechanistic aspects of DNA transposition [J]. Current Opinion in Genetics & Development, 1992, 2(5):698-704.
