曲古抑菌素A对缺糖/缺氧损伤PC12细胞的保护作用
2013-12-07越茂松王进京季秋虹季煜华
越茂松,黄 琼,王进京,季秋虹,季煜华,3
(1.暨南大学 生命科学技术学院 组织移植与免疫研究中心广州 510632,2.南通大学附属医院神经内科,南通 226001,3.暨南大学功能蛋白质研究广东普通高校重点实验室广州 510632)
缺血性脑卒中是世界范围内导致成年死亡和残疾的重要原因,但目前除了溶栓治疗外还缺乏其他理想的治疗药物和手段[1]。乙酰化是一种重要的表观遗传调控机制,组蛋白乙酰化转移酶(Histone acetyltransferases,HATs)和组蛋白去乙酰化酶(Histone deacetylases,HDACs)维持和调节着机体中蛋白质乙酰化水平的动态平衡[2]。近年来,一系列的报道证实了卒中以及多种神经退行性疾病(HD,
Alzheimer等)都伴随着HATs蛋白量/活性的下降和HDACs活性的相对增加,而通过组蛋白去乙酰化酶抑制剂(Histone deacetylase inhibtor,HDACis)抑制HDACs的活性,增加乙酰化水平可降低神经元损伤的程度并促进其功能的恢复[3-5]。曲古抑菌素A(TSA)是一种泛去乙酰化酶抑制剂,通过改变染色质折叠构象调节基因表达和改变蛋白质乙酰化水平调节蛋白质的功能。[4,6]本研究拟检测 TSA对 PC12细胞OGD损伤模型的作用并探讨可能的机制。
1 材料与方法
1.1 主要材料与仪器 PC12,ATCC;TSA,碧云天生物技术研究所;DMEM无糖培养基,美国Sigma公司;DMEM高糖培养基、胎牛血清和马血清,美国Gibco公司;MTT,美国 Sigma公司;DCFH-DA,碧云天生物技术研究所;超净工作台(水平流型),中国ESCO公司;CO2细胞培养箱(2300型),美国公司Sheldon公司;倒置显微镜(COICXSZ-D2型),日本尼康公司;680型酶标仪,美国Bio-Rad公司;荧光显微镜,日本尼康公司;流式细胞仪,美国BD公司。
1.2 实验方法与步骤
1.2.1 PC12细胞培养 细胞复苏后接种于用多聚赖氨酸包被过的培养瓶中,用含5%胎牛血清(FBS)和10%马血清(HS)的高糖DMEM完全培养基在5%CO2,37℃条件下恒温、恒湿培养箱中培养,细胞汇合度70% ~80%时按1∶4传代。
1.2.2 MTT检测TSA对细胞增殖的影响 按1×104个细胞/孔、200 μl接种用多聚赖氨酸包被过的96孔板,在5%CO2、37℃恒温恒湿培养箱培养24 h后,换成含不同浓度TSA的完全培养基继续培养,处理 48 h 后,每孔加入5 g·L-1MTT 20 μl,再培养4 h,每孔加入三联裂解液150 μl过夜,待孔内颗粒完全溶解后,用酶标仪,选择波长570 nm,检测各孔吸光度(A),抑制率%=1-A实验组/A对照组×100%,实验重复3次,结果做统计分析。
1.2.3 模型的建立及实验分组 参考文献方法稍加改进建立缺糖/缺氧模型,取处于对数生长期的PC12细胞按1×104个/孔、200 μl接种于用多聚赖氨酸包被过的96孔板中,在5%CO2、37℃恒温恒湿培养箱培养24 h,将其分为 blank control、model control和OGD组,每组设6个复孔。OGD组设六个浓度,分别加入含 CoCl2为 25、50、100、200、400、800 μmol·L-1的无糖 DMEM 培养基 100 μl,37℃,5%CO2培养2 h,用MTT法检测细胞存活率,确定最佳造模浓度。
1.2.4 利用PI和Hochest染色检测细胞凋亡与坏死 取处于对数期的PC12细胞按1×105个 /孔、600 μl接种于用多聚赖氨酸包被过的24孔板,分组,细胞贴壁过夜,将各组旧培养基小心吸出后,用PBS洗两遍,将其分为blank control、model control和OGD组。OGD组设3个浓度,分别加入含CoCl2为50、200、800 μmol·L-1的无糖 DMEM 培养基300 μl,37℃,5%CO2培养2 h,小心吸干上清后,用PBS洗两遍,加入0.01 g·L-1Hoechst 33258和0.01 g·L-1PI(用PBS溶液配制成0.01 g·L-1)各100 μl,避光反应10 min吸弃上清,用PBS洗两遍后,加入含10%FBS的PBS 200 μl,荧光显微镜观察摄像。PI荧光为红色(620 nm),Hoechst 33258荧光为蓝色(480 nm)。
1.2.5 MTT法检测细胞存活率 按1×104个细胞/孔、200 μl接种用多聚赖氨酸包被过的96孔板,分组,将其分为control组,OGD组,给药组。给药组设7 个浓度,分别为:1、10、40、80、160、320、640 nmol·L-1。每组设 6个复孔,在 5%CO2、37℃恒温恒湿培养箱培养24 h后,将各组旧培养基吸出后control组加入100 μl完全培养基,给药组含不同浓度TSA的完全培养基继续培养4 h,将除control外的其他各组的旧培养基小心吸出后,用PBS洗两次,OGD 组加入含 CoCl2100 μmol·L-1的无糖 DMEM培养基100 μl,给药组加入含不同浓度TSA的OGD培养液100 μl,37 ℃,5%CO2培养箱培养2 h后,每孔加入终浓度为 5 g·L-1的 MTT 溶液 10 μl,继续培养4 h后,每孔加入三联裂解液150 μl过夜,待孔内颗粒完全溶解后,用酶联免疫检测仪,选择波长570 nm,检测各孔吸光度(A)。
1.2.6 检测细胞内活性氧含量
1.2.6 .1 荧光显微镜检测细胞内活性氧含量 取对数生长期的PC12细胞消化收集并接种于24孔板中,贴壁过夜。分别加药处理,并设阳性对照组和阴性对照组。给药组造模前提前4 h加入不同浓度TSA处理,2 h后小心吸去细胞培养液,每孔加入200 μl按照1 ∶1000用 PBS稀释好的DCFH-DA,使终浓度为 10 μmol·L-1。37℃细胞培养箱内孵育20 min。用PBS洗涤细胞3次,以充分去除未进入细胞内的DCFH-DA。最后每孔加入500 μl PBS,阳性对照组加入1 μl活性氧阳性对照刺激物Rosup。20 min后用荧光显微镜在波长485 nm激发光下观察。
1.2.6 .2 流式细胞仪检测细胞内活性氧含量 取对数生长期的PC12细胞消化收集并接种于6孔板中,贴壁过夜。分别加药处理,并设阳性对照组和阴性对照组。给药组造模前提前4 h加入不同浓度TSA处理,2 h后收集细胞到相应流式管中,每管加入 400 μl终浓度为 10 μmol·L-1的 DCFH-DA,37℃细胞培养箱内孵育20 min用PBS洗涤细胞3次,以充分去除未进入细胞内的DCFH-DA。最后每孔加入500 μl PBS,阳性对照组加入1 μl活性氧阳性对照刺激物Rosup。20 min后用流式细胞仪检测。
2 结果
2.1 TSA对PC12细胞增殖活性的影响 随着TSA浓度升高,细胞的增殖活性逐渐降低,从5 nmol·L-1起明显抑制增殖活性(P<0.05),并在160 nmol·L-1时抑制50%的增殖活性,见Fig 1。
2.2 PC12细胞缺糖/缺氧损伤模型的建立 实验结果表明随着CoCl2浓度的增加细胞存活率相应下降。实验选择PC12细胞OGD损伤2 h,存活率为61%时 CoCl2浓度为 100 μmol·L-1作为下一步实验造模浓度。见Fig 2。

Fig 1 Survival rate of PC12 cell with TSA for 48 h( ± s)

Fig 2 Survival rate of PC12 cell with CoCl2for 2 h(n=6)
2.3 利用PI和Hochest染色检测细胞凋亡与坏死实验观察随着CoCl2浓度的增加,与blank control相比,细胞凋亡与坏死的数量不断增加,尤其是800 μmol·L-1CoCl2组最为明显。结果见Fig 3。

Fig 3 Cell apoptosis and necrosis observed in PI and Hoechst staining(10×10)
2.4 TSA对缺糖/缺氧损伤的PC12细胞存活率的影响 MTT法检测显示,10~160 nmol·L-1TSA组全程作用6 h后,经缺糖/缺氧损伤的PC12细胞存活率与模型组相比差异有显著性(P<0.05),特别是80 nmol·L-1的TSA组与OGD组相比其存活率提高了29%。结果见Fig 4。
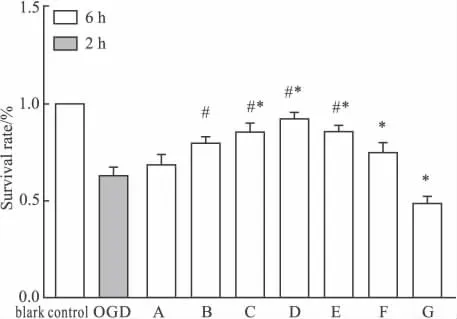
Fig 4 Effect of TSA on survival rate of OGD on PC12 cells( ± s)
2.5 荧光显微镜检测PC12细胞活性氧的含量采用DCFH-DA荧光探针对细胞内活性氧含量进行测定。实验结果显示PC12细胞经OGD损伤2 h后,ROS生成量明显增加;TSA全程处理的PC12细胞OGD损伤2 h后,细胞内ROS生成量与OGD处理组比较明显减少,特别是80 nmol·L-1TSA+OGD组。实验结果表明TSA能够拮抗OGD损伤造成的PC12细胞内ROS的增加。结果见Fig 5。

Fig 5 Effect of TSA on oxygen content of active cells(10×10)
2.6 流式细胞仪检测PC12细胞内活性氧含量采用DCFH-DA荧光探针对细胞内活性氧含量进行测定。流式细胞仪显示的数据与荧光显微镜的结果吻合。PC12细胞经OGD损伤2 h后,ROS生成量明显增加,TSA全程处理的PC12细胞OGD损伤2 h后,细胞内ROS生成量与OGD组相比较明显减少,特别是80 nmol·L-1TSA+OGD组。由此证明80 nmol·L-1TSA+OGD能明显拮抗OGD损伤造成的PC12细胞内ROS的增加。结果见Fig 6。

Fig 6Effect of TSA on oxygen content of active cells(±s)
3 讨论
目前认为神经元损伤是缺血性脑血管疾病的发病机制中的重要因素,损伤涉及多方面,其包括能量代谢障碍、ROS损伤、神经元细胞内电解质失衡(钙、钠离子超载)、脂质过氧化反应、线粒体损伤及细胞凋亡等。[7-9]在这些诸多的损伤因素中,能量代谢障碍可能是首发环节,因此缓解细胞能量代谢障碍被认为是治疗缺血性卒中的一个重要的治疗靶点[10]。
实验运用CoCl2合并无糖培养基模拟细胞缺血损伤过程,建立了体外神经元的缺血模型。[11]发现100 μmol·L-1CoCl2合并缺糖 2 h,细胞存活率为61%,且细胞无明显的坏死,因此适宜用此浓度建模。通过大量MTT前期实验筛选出PC12细胞添加TSA的最佳方案,即通过TSA对PC12细胞预处理4 h,OGD过程也同时加入TSA(全程加入法)、TSA只对PC12细胞预处理4 h(预处理法)和OGD过程同时加入TSA(同时加入法)三种处理方案的MTT数据比对,发现全程加入法对细胞的保护效果最佳。
近期文献报道,细胞中参与糖酵解,糖异生,三羧酸循环,尿素循环,脂肪酸循环,糖原代谢中的多种酶都会被乙酰化修饰,并且细胞外能量物质的浓度将直接影响这些代谢过程中酶的乙酰化程度[12,13]。然而近期有文章指出,许多参与糖酵解和脂肪代谢的酶的乙酰化程度直接影响其活性,乙酰化程度高,则酶活性强。已经证实:GADPH的乙酰化加速糖酵解的反应,增加烯酰-COA水合酶和3-L-羟脂酰-COA的乙酰化水平也促进脂肪代谢,最终产生AC-COA,进入三羧酸循环(TCA)产生的NADH,FADH2再进行氧化磷酸化反应产生大量 ATP[10]。MTT结果显示,与 OGD组相比,80 nmol·L-1TSA预处理PC12细胞4 h可明显提高细胞的存活率。由于TSA处理时间较短(4 h),因此有可能是通过增加能量代谢过程中一些关键酶的乙酰化修饰,从而增加或维持细胞内能量的释放,以应对因OGD引起的能量代谢障碍,进而提高细胞的存活率,达到保护细胞的作用。
脑缺血造成的能量代谢障碍将促使细胞内产生大量的ROS,它们与脂质、蛋白质、及核酸发生反应引起膜脂质过氧化,导致膜损伤,线粒体功能障碍,细胞溶解和组织水肿等一系列损害作用[8]。研究结果显示80 nmol·L-1TSA预保护组的胞内ROS生成量明显下降。因此,TSA对OGD损伤PC12细胞的保护作用,可能通过增加能量代谢酶的乙酰化水平,缓解缺血引起的能量障碍,进而降低细胞OGD损伤引起的ROS生成量,但具体的调控机制还有待进一步研究。
[1]Donnan G A,Fisher M,Macleod M,et al.Stroke[J].Lancet,2008,371(9624):1612-23.
[2]Saha R N,Pahan K.HATs and HDACs in neurodegeneration:a tale of disconcerted acetylation homeostasis[J].Cell Death Differ,2006,13(4):539-50.
[3]Kim H J,Leeds P,Chuang D M.The HDAC inhibitor,sodium butyrate,stimulates neurogenesis in the ischemic brain [J].J Neurochem,2009,110(4):1226-40.
[4]Kazantsev A G,Thompson L M.Therapeutic application of histone deacetylase inhibitors for central nervous system disorders[J].Nat Rev Drug Discov,2008,7(10):854-68.
[5]Langley B,Brochier C,Rivieccio M A.Targeting histone deacetylses as a multifaceted approach to treat the diverse outcomes of stroke[J].Stroke,2009,40(8):2899-905.
[6]Mohammmad Shahidul Makki,Thorsten Heinzel,Christoph Englert.TSA downregulates Wilms tumor gene 1(Wt1)expression at multiple levels[J].Nucleic Acids Research,2008,36(12):4067-78.
[7]宋文婷,徐 立,刘建勋.脑缺血后谷氨酸及其受体介导的神经细胞损伤及相关药物研究进展[J].中国药理学通报,2012,28(6):747-50.
[7]Song H,Wang M X,Xu R A.Role of plasminogen system in myofibroblast apoptosis and tissue fibrosis[J].Chin Pharmacol Bull,2012,28(6):747-50.
[8]安泳潼,夏玉叶,闵 旸.缺血性脑卒中的发病机制及其治疗[J].世界临床药物,2010,31(1):35-9.
[8]An Y T,Xia Y Y,Min Y.The pathogenesis of cerebral ischemia and its treatment[J].World Clini Drugs,2010,31(1):35-9.
[9]宋修云,胡金凤,陈乃宏.神经细胞凋亡与脑缺血疾病[J].中国药理学通报,2012,28(3):307-10.
[9]Song X Y,Hu J F,Chen N H.Neurons apoptosis and cerebral ischemia[J].Chin Pharmacol Bull,2012,28(3):307- 10.
[10]Leonard Guarente.The Logic Linking Protein Acetylation and Metabolism[J].Cell Metabolism,2011,14(2):151-3.
[11]William J Goldberg,Richard M.Kadingo,et al.Effects of Ischemialike Conditions on Cultured Neurons:Protections:Protection by Low Na+,Low Ca2+Solutions[J].J Neurosci,1986,6(11):3144-51.
[12]Shimin Zhao,Wei Xu,Wenqing Jiang,et al.Regulation of Cellular Metabolism by Protein Lysine Acetylation[J].Science,2010,327(5968):1000-4.
[13]Qijun Wang,Yakun Zhang,Chen Yang,et al.Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux[J].Science,2010,327(5968):1004-7.
