TNFR1-383A/C基因多态性在散发性帕金森病患者中的分布及临床特点分析
2013-11-23周涌涛刘佳张燕莉张新卿郭秀海陈彪贾建平
周涌涛 刘佳 张燕莉 张新卿 郭秀海 陈彪 贾建平
帕金森病(PD)是老年人群中常见的神经系统变性疾病,尽管其发病机制尚不清楚,但越来越多的证据表明遗传因素和免疫机制参与了疾病的发生。研究发现PD患者脑组织中存在活化的小胶质细胞,并广泛表达细胞因子等炎性物质,且患者脑脊液中肿瘤坏死因子α(TNFα)等细胞因子水平升高,流行病学研究发现长期应用非甾体药物可减少 PD 的 发 生[1-3]。TNFα 与 TNF 受 体 1(TNFR1)相互作用可产生兴奋性毒性物质,导致线粒体结构异常,黑质神经元凋亡[4-7]。目前有关其受体多态性与PD之间关系的研究较少。一项来自德国人群的研究发现TNFRSF-609和+36位点可能为PD的保护性因素[8-11],近来研究发现TNFα的水平与PD患者非运动症状相关,如可影响患者的认知功能、睡眠及情绪等[12]。本研究采用病例对照的研究方法,探讨了TNFR1-383A/C基因多态性与PD之间的关联性,并探讨该基因多态性对PD发病年龄及运动症状的影响。
1 对象和方法
1.1 对象
1.1.1 PD组:共518例,来自宣武医院帕金森病专科门诊以及全国多中心的药物实验(北京、成都、上海、天津、长沙、武汉、青岛及哈尔滨)。入组标准:(1)符合 PD 诊断[13];(2)年龄≥50岁;(3)无PD家族史;(4)经过1年以上随访。排除标准:(1)具有其他神经系统变性疾病;(2)患有脑血管病;(3)有精神系统疾病。平均发病年龄(61.35±8.34)岁,确诊时平均年龄(65.03±7.92)岁,其中男317例、女201例。所有患者均无遗传关联性,获得患者本人知情同意并签订知情同意书。
1.1.2 对照组:共521名,从北京老年化纵向研究的数据库(数据库共2200名北京社区人群,收集日期为2005-04-2009-06)中随机抽取,入组者第1次随访年龄在50岁以上且病史中无神经系统疾病者,共随访4年。
1.2 方法
1.2.1 实时定量荧光 PCR(real time PCR,RTPCR)[9]:(1)应用氯仿抽提的方法提取外周血中的DNA;(2)引物和Taq酶由Roche公司提供,应用RT-PCR[9]进行检测;两对引物序列为:第1对:上游 5′-TCCAGGACTGCATGGAC-3′和 下 游 5′-AAAAGTCCTTACCTCGGCAGT-3′,第2对:上游5′-GTCCAGGACTGCATGGAT-3′,下 游 5′-AAAAGTCCTTACCTCGGCAGT-3′;(3)RTPCR扩增的总体积为50μL,其中含有deltZ05、10×deltZ05缓冲液、10mmol/L dNTP、200μmol/L ROX、20×SYBR Green、80%(体积分数)甘油、100%DMSO。PCR反应条件:95℃初变性12 min,然后95℃20s,58℃退火20s,45个循环后,从60℃进行解离20min。实验操作流程和数据分析通过罗氏公司抽查和重复测试,进行质控。
1.2.2 TNFR1-383A/C 基因多态性与 PD 的相关性:分析TNFR1-383A/C基因型和等位基因频率在不同性别、发病年龄以及有无震颤者等之间的差异。
1.3 统计学处理 数据分析采用SPSS 11.5统计软件,应用Mantal-Haenszelχ2检验对计数资料进行分析,应用非条件Logistic回归模型进行多因素分析,并计算OR 和95%CI。应用 Kaplan-Meier、life table和 Cox-regression方法分析基因多态性对发病年龄的影响。以P<0.05为差异有统计学意义。
2 结果
2.1 TNFR1-383A/C基因型和等位基因频率TNFR基因型的分布符合Hardy-Weinberg平衡(P<0.05)。病例组和对照组CC、CA和AA基因型分布无统计学差异(P>0.05),C和A等位基因频率在两组间比较亦无统计学差异(P>0.05)。PD患者发病年龄≥65岁亚组C等位基因频率显著低于对照组≥65岁亚组(6.74%vs.10.10%,P<0.05),并可减少晚发型PD发病的危险性(OR:0.64,95%CI:0.43~0.97,P<0.05)。两组同性别间未见C等位基因频率存在统计学差异(均P>0.05)。具体结果见表1。
把病例和对照组进行年龄匹配,利用非参数Logistic回归模型对性别进行调整,未发现C/C基因型可影响SPD发病的危险性(OR:0.55,95%CI:0.13~2.33,P=0.418),也未发现 TNFR1-383A/C/C等位基因对SPD发病有显著影响(OR:0.85,95%CI:0.61~1.19,P=0.347)。
2.2 TNFR1-383A/C基因多态性对临床特征的影响 震颤组和非震颤组CC、CA和AA基因型的频率以及C和A等位基因的频率与对照组比较均无统计学差异(P>0.05)(表1)。
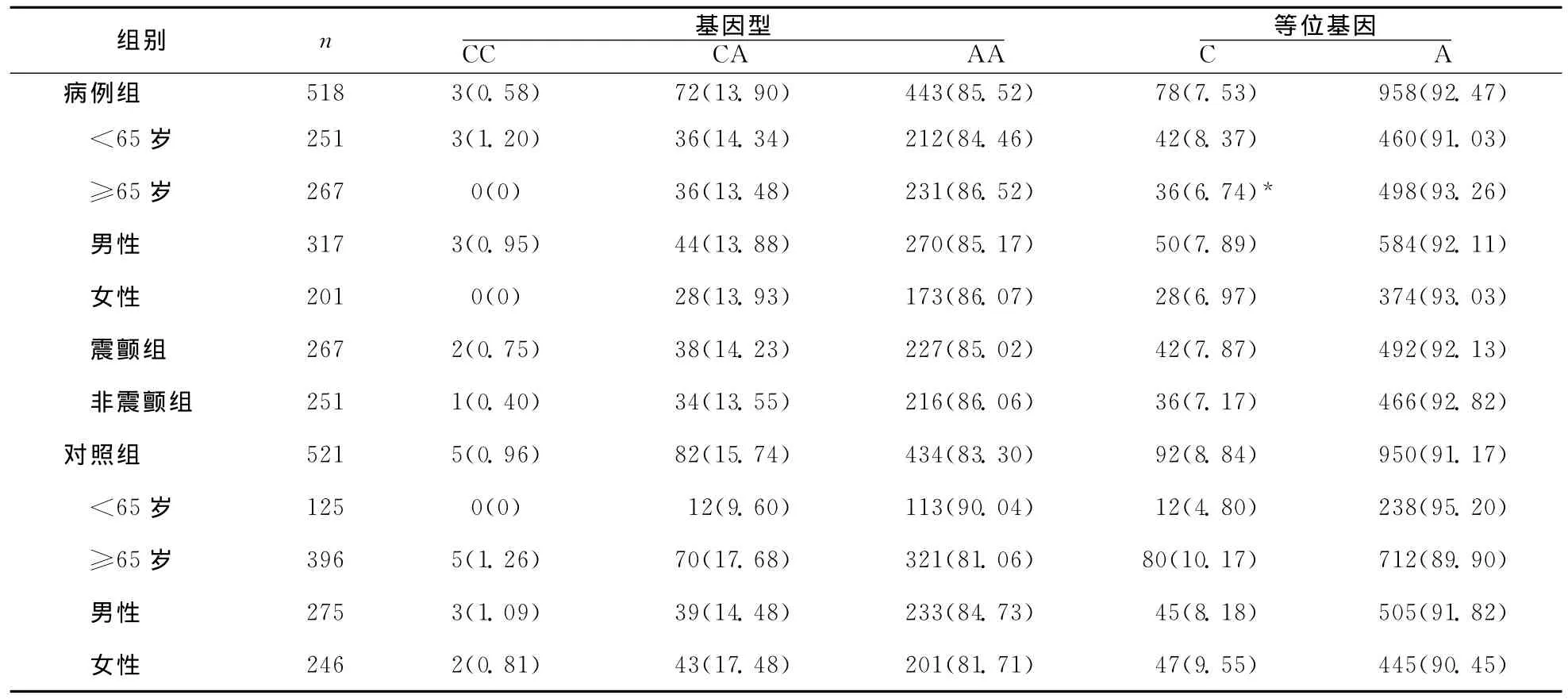
表1 对照组和病例组TNFR1基因型和等位基因的分布 〔n(%)〕
2.3 利用生存分析确定基因多态性对发病年龄的影响 CC、CA和AA基因型患者的发病年龄的中位数分别为54岁(95%CI:46~62岁)、62岁(95%CI:58~66岁)和61岁(95%CI:60~62岁),各基因型患者间比较无统计学差异(P=0.16),携带C/C等位基因的个体并未延迟SPD发病年龄。携带C和非C等位基因个体发病年龄的中位数分别为61岁(95%CI:58~64岁)与61岁(95%CI:60~62岁),两者间无统计学差异。利用生存分析进行研究发现携带C等位基因的个体并不能延迟SPD的发病年龄(C allele vs.non-C allele log rankχ2=0.22,P=0.86)。
3 讨论
TNFR1基因位于12p13.31,包含10个外显子,编码TNFR1蛋白的相对分子质量为55000~60000。TNFR1属于跨膜蛋白受体,膜外包括富含半胱氨酸的区域,有核细胞上均有低水平表达,包括大脑的各个区域,参与了TNF作用的调节,如成纤维细胞生长、内皮细胞激活或黏附、抗病毒、非淋巴细胞进行的细胞毒性作用和凋亡。在PD患者黑质多巴胺能神经元内发现TNFR1表达水平升高,但TNFR2的水平没有改变,并在动物模型中发现TNFR1对神经元具有毒害作用,而TNFR2则具有保护作用[4-6,14]。对于 TNFR1基因位点-609和+36位点的研究发现其可能为PD发病的保护性因素,但目前尚未发现有关TNFR1-383A/C多态性与SPD之间关系的研究报道。
本研究结果显示,TNFR1-383A/C基因型和等位基因在病例组和对照组之间未存在统计学差异,病例组CC基因型频率低于对照组,并未发现CC基因型和C等位基因可影响SPD发病的危险性,通过对性别和年龄调整之后仍未发现两者间具有相关性。但按照性别和年龄分层分析后发现,高龄PD患者(发病年龄≥65岁)C等位基因频率显著低于对照组,并可减少晚发型PD发病的危险性,表明在发病年龄≥65岁人群中C等位基因仍是SPD的保护性因素,提示TNFR1-383A/C基因多态性与SPD的保护性作用有关。进一步分析TNFR1-383A/C对发病年龄的影响发现,具有C和A等位基因者的发病年龄无统计学差异,表明C等位基因并不能影响SPD的发病年龄。在临床工作中发现PD患者的起病形式有的以震颤为主,而许多患者并无震颤,而以运动迟缓和肌强直为主。本研究并未发现TNFR1-383A/C基因型和等位基因与SPD的临床表现存在相关性,表明该位点的多态性对SPD的运动症状并未产生影响。本研究与德国研究均发现该位点可能对SPD的发病具有保护性作用[8-11]。其机制可能为该突变位点可能通过影响TNF-αmRNA水平和表观遗传学机制调节TNF-α体内的水平,从而影响SPD的病理过程[14]。本研究在基因型和等位基因频率与其他研究结果之间存在差异[15-16],其原因主要考虑与种族差异、地理区域以及饮食习惯不同等因素有关,也不能完全排除本研究中的病例对照区域不匹配导致。
综上所述,TNFR1-383A/C可能与SPD高龄人群的发病存在相关性,这与既往的动物实验、流行病学研究以及脑脊液生化研究的结果存在不同,因而需要通过对其他易感位点进行深入研究以确定其与SPD的关系。
[1]Arai H,Furuya T,Mizuno Y,et al.Inflammation and infection in Parkinson’s Disease[J].Histol Histopathol,2006,21:673-678.
[2]Lucas SM,Rothwell NJ,Gibson RM.The role of inflammation in CNS injury and disease[J].Brit J Pharmacol,2006,147:S232-S240.
[3]Chen H,Jacobs E,Schwarzschild MA,et al.Nonsteroidal antiinflammatory drug use and the risk of Parkinson’s disease[J].Ann Neurol,2005,59:988-989.
[4]Hirsch EC,Breidert T,Rousselet E,et al.The role of glial reaction and inflammation in Parkinson’s disease[J].Ann N Y Acad Sci,2003,991:214-228.
[5]Nagatsu T,Sawada M.Inflammatory process in Parkinson’s disease:role for cytokines[J].Curr Pharm Des,2005,11:999-1016.
[6]Nagatsu T,Sawada M.Cellular and molecular mechanisms of Parkinson’s disease:neurotoxins,causative genes,and inflammatory cytokines[J].Cell Mol Neurobiol,2006,26:781-802.
[7]Whitton PS.Inflammation as a causative factor in the aetiology of Parkinson’s disease[J].Br J Pharmacol,2007,150:963-976.
[8]Tansey MG,Frank-Cannon TC,McCoy MK,et al.Neuroin-flammation in Parkinson’s disease:is there sufficient evidence for mechanism-based interventional therapy?[J].Front Biosci,2008,13:709-717.
[9]Nishimura M,Mizuta I,Mizuta E,et al.Tumor necrosis factor gene polymorphisms in patients with sporadic Parkinson’s disease[J].Neurosci Lett,2001,311:14.
[10]Wu YR,Feng IH,Lyu RK,et al.Tumor necrosis factor-alpha promoter polymorphism is associated with the risk of Parkinson’s disease[J].Am J Med Genet B Neuropsychiatr Genet,2007,144B:300-304.
[11]Krüger R,Hardt C,Tschentscher F,et al.Genetic analysis of immunomodulating factors in sporadic Parkinson’s disease[J].J Neural Transm,2000,107:553-562.
[12]Menza M,Dobkin RD,Marin H,et al.The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease[J].Psychosomatics,2010,51:474-479.
[13]Gelb DJ,Oliver E,Gilman S.Diagnostic criteria for Parkinson disease[J].Arch Neurol,1999,56:33-39.
[14]Nagatsu T,Sawada M.Cellular and molecular mechanisms of Parkinson’s disease:neurotoxins,causative genes,and inflammatory cytokines[J].Cell Mol Neurobiol,2006,26:781-802.
[15]Nishimura M,Obayashi H,Mizuta I,et al.TNF,TNF receptor type 1,and allograft inflammatory factor-1gene polymorphisms in Japanese patients with type 1diabetes[J].Hum Immunol,2003,64:302-309.
[16]Bridges SL Jr,Jenq G,Moran M,et al.Single-nucleotide polymorphisms in tumor necrosis factor receptor genes:definition of novel haplotypes and racial/ethnic differences[J].Arthritis Rheum,2002,46:2045-2050.
