儿童激素耐药肾病综合征相关致病基因及其临床检测策略
2013-11-22李国民
李国民 沈 茜 徐 虹 安 宇
肾病综合征(NS)是因肾小球滤过屏障受损而引起的一组临床综合征。肾小球滤过屏障由内皮细胞、基底膜和足细胞构成。多项研究表明,足细胞在肾小球滤过屏障中起关键作用,特别是足细胞相邻足突构成的裂隙膜(slit diaphragm,SD)[1~4]。依据对激素治疗的反应可将 NS 分为激素敏感型(SSNS)和激素耐药型 (SRNS)[5]。10%~20%的NS患儿为SRNS,超过半数的SRNS患儿在10年内会进展为终末期肾病(ESRD)[5~7]。在 NS患者中,有一类是由足细胞和基底膜相关分子基因突变所引起,该类NS又称为遗传性NS[8,9]。遗传性NS患者不仅表现对激素耐药,对其他免疫抑制剂几乎均耐药,属于SRNS范畴,治疗的主要方向是减轻蛋白尿和保护肾功能,延缓疾病进展;这类SRNS患者易快速进展为ESRD,而进展为ESRD后的最佳治疗选择为肾移植,因为遗传性NS患者肾移植后复发率很低[10~13]。目前临床观点认为,在SRNS患者选择免疫抑制剂治疗和终末期肾移植之前应将遗传性NS患者筛选出来,其目的是为SRNS患者提供个体化治疗,避免盲目治疗[14~17]。本文就SRNS相关致病基因、主要基因突变流行率,及临床基因分析对象、方法和意义进行综述。
1 SRNS的定义和分类
儿童SRNS诊断标准:泼尼松1.5~2.0 mg·kg-1·d-1(最大不超过60 mg)治疗4~8周,或相应剂量的其他类型的肾上腺皮质激素治疗4~8周,尿蛋白不能转阴[18,19]。依据SRNS患者是否伴有肾外症状,可将 SRNS分为非综合征型(单发型)SRNS 和综合征型 SRNS[8,14]。非综合征型SRNS只有NS表现,不伴有其他系统症状,综合征型SRNS不仅有NS的表现,还有先天性或遗传性疾病引起的肾外症状;依据发病年龄又可以将SRNS分为先天性(0~3个月)、婴儿型(~12个月)、儿童早发型(~5岁)、儿童迟发型(~13岁)、青少年型(~18岁)和成人型(>18 岁)[8,14];依据患者间是否具有血缘关系,SRNS 可分为家族性和散发性。
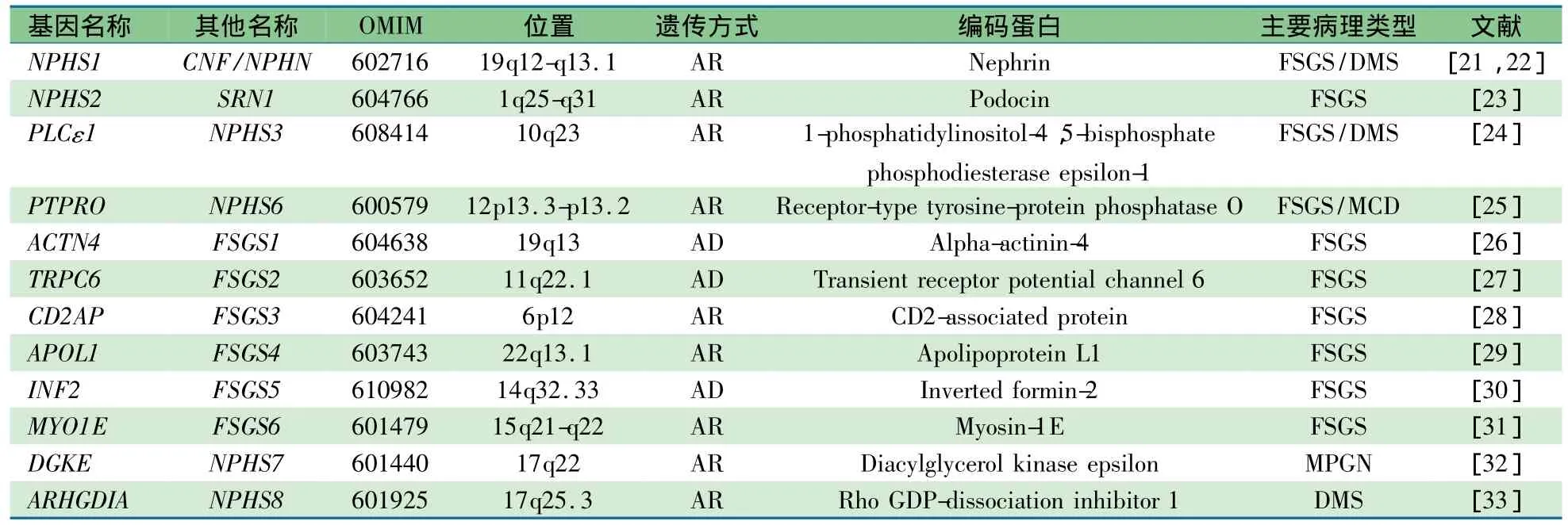
表1 非综合征型SRNS及其致病基因
2 SRNS的相关致病基因
SRNS患者发病早,有肾病水平蛋白尿家族史,快速进展为 E SRD 提 示由遗传因素引起[14,15,20]。1998 年,发现了NPHS1,其编码nephrin是足细胞SD结构构成和信号传导分子,是维持肾小球滤过屏障完整性的关键分子之一,也是芬兰型先天性NS主要致病基因[21]。NPHS1的发现开创了NS致病基因和分子研究时代。
2.1 非综合征型(单发型)SRNS致病基因 目前有12种基因突变可分别引发非综合征型SRNS(表1),其临床主要病理类型为局灶节段肾小球硬化(FSGS)、弥漫系膜硬化(DMS)和膜增生性肾小球肾炎(MPGN)。这些基因产物均在肾小球足细胞中表达,有的分子直接、间 接 参 与 SD(NPHS1、NPHS2、CD2AP)和肌动蛋白细胞骨架(ACTN4、INF2)的构成,有的参与细胞代谢和信号转导(PTPRO、 PLCE1、 APLO1、MYO1E、DGKN 和 ARHGDIA)[20~32]。 N PHS1、 N PHS2、PTPRO、PLCE1、CD2AP、APLO1、MYO1E、DGKN 和 A RHGDIA引起的SRNS表现为常染色体隐性遗传(AR),在儿童时期发病(APLO1除外,其突变主要在成年期发病);而ACTN4、TRPC6和INF2引起的SRNS表现为常染色体显性遗传(AD),多数患者在青少年或成年期发病[12,14,34]。APLO1与成人ESRD、FSGS和非糖尿病肾病发病有关,这些疾病主要发生在非裔美国人群中,未见其在其他人群中发生,表明APLO1所致疾病与种族有关联[29,35,36]。这些基因突变类型有错义、无义突变、插入、缺失和剪切突变等,其中错义突变是主要突变类型,可以累及基因所有编码区,故无热点突变[14,15,37]。
2.2 综合征型SRNS致病基因综合征型SRNS患者肾外症状明显,经常因肾外症状就诊,后发现肾脏表现。引起综合征型SRNS的基因见表2,这些基因不仅仅在足细胞中表达,而且在其他组织和细胞也有表达。其中WT1和LMX1B编码的蛋白属于转录因子,LAMB2和ITGB4编码的蛋白属于肾小球基底膜构成分子,SCARB2编码的蛋白属于溶 酶 体 蛋 白 ,COQ2、PDSS2、COQ6和MTTL1编码的蛋白属于线粒体蛋白,SMARCAL1编码的蛋白属于DNA核小体重组调节因 子[37~52]。WT1、LAMB2 和MYH 9突变可仅有SRNS表现(非综合征型),也可引起相应的综合征同时伴发SRNS(综合征型)[40,54,55]。WT1、MYH9、ITGB4、ZMPSTE24 和 LMNA突变可以引发多个临床综合征,并不是都伴有SRNS,仅部分伴有SRNS(表2)。如WT1突变可以引起中疾病,Denys-Drash综合征和Frasier综合征可伴有NS表现,而体细胞间皮瘤和Wilms肿瘤1型无NS表现。此外,WT1突变也可仅有NS表现,所以有时 WT1可用 NPHS4作为基因代码[54,56,57]。Galloway-Mowat综合征于1968首次描述,临床表现为早期发病的NS(病理DMS)、小头畸形和裂孔疝,为AR,至今尚未发现其致病基因[58]。

3 SRNS相关致病基因的突变频率
澳大利亚的研究发现,SRNS是儿童继先天性肾脏泌尿道畸形(CAKUT)之后第二常见的遗传性肾脏疾病[59]。在综合征型SRNS致病基因中,WT1是所致疾病相对常见的基因,其他基因所致疾病比较罕见[38]。在非综合征型SRNS致病基因中,PTPRO所致疾病不太常见,可能与相关研究不够有关[15]。国外研究显示,相对常见的SRNS相关致病基因在不同人群的流行率是不同的(表3)[9,12,14,60~68]。国 内 对 SRNS 致 病 基 因 研 究 比 较 少,NPSH2在散发性SRNS患儿中的突变率为4%~5%[69],WT1在3岁之前散发性SRNS患儿中的突变率为16%~17%,3岁之后未见报道[70]。NPHS1在散发性SRNS患儿中的突变率为4.5%[71]。世界各地研究发现,某一基因的突变率并不一致,可能是种族的影响。总体特征是,年龄越小发病的 SRNS,基因所致可能性越大[15,72~74]。

表310个相对常见SRNS基因的突变率(%)
4 SRNS相关致病基因分析对象和策略
4.1 分析对象 从SRNS相关致病基因在不同人群中流行特点来分析,儿童是基因分析的主要对象,无论是散发性还是家族性 S RNS 均应该进行相关基因分析[14,15,60]。NPHS2、ACTN4、TRPC6和INF2在家族性SRNS成人病例中有一定的突变率,但在散发性SRNS成人中罕见,因此家族性AR的 SRNS患者可行 NPSH2分析,AD患者可行ACTN4、TRPC6和INF2分析,散发性SRNS成人病例可不行基因分析[9,16,34]。
4.2 分析方法 目前,SRNS相关致病基因分析方法主要是用Sanger法对外显子及附近区域直接测序,并已经从实验室走向临床[14,15,60]。这种方法的优点是准确性高、重复性强,缺点是需要消耗大量的财力和时间,这也限制了SRNS相关致病基因分析在临床推广。对于有热点突变的基因,基于SnapShot等技术的热点突变分析是目前临床经济、有效和快速的基因分析方法。随着科技的发展,高通量测序仪已在生物科学领域应用。基于芯片技术的多基因外显子捕获和高通量测序将是未来SRNS相关致病基因全面分析的方向[75]。
4.3 分析策略 涉及SRNS发病基因达到数十个,临床对所有SRNS致病基因进行分析无疑是很昂贵和耗时、也不切实际。临床理想的SRNS致病基因分析策略是根据表型与基因型的对应关系,有针对性的进行某些基因分析,但SRNS患儿表型与基因型具有异质性,非对应关系,这就给临床SRNS患者基因分析带来困难。对于综合征型SRNS患儿,致病基因分析的策略是根据肾外表现来推断可能的致病基因,然后对该基因进行直接测序(图1)[9,11,20,42,46,60,76]。而对于非综合征 S RNS 患儿,目前临床策略是对非综合征SRNS患儿先行肾活检,依据病理表型来推断可能的致病基因,然后进行候选致病基因直接测序(图 2 )[8,15,16,60,77],这样可尽可能缩小检测基因范围。但近年来有学者认为,SRNS患儿致病基因分析应该先于肾活检,其理由为肾活检具有创伤性,肾脏病理类型与致病基因并没有直接的关系。尽管多数SRNS患儿主要肾脏病理为FSGS和DMS。但研究发现,遗传性SRNS患儿的早期肾脏病理改变可为轻微病变(MCD)或系膜增殖性肾小球肾炎(MsPGN),晚期可以表现为FSGS或DMS,并无特征性改变[14,15]。由于1岁以内儿童 N S主要由 N PHS1、NPHS2 和WT1等基因突变引起,特别是先天性肾病综合征(CNS)[74,78]。因此,对 C NS 和婴儿型 N S 患儿应常规行NPHS1、NPHS2和 WT1等基因分析,更应该先于肾活检[8,15,60,78]。合理的儿童 S RNS 相关致病基因临床分析策略应该是根据特定SRNS儿童人群(遗传背景相似)的突变图谱来选择致病基因分析。这就需要通过对特定SRNS儿童人群进行致病基因研究,建立该人群SRNS致病基因突变图谱。成人主要依据是家族性或散发性来决定基因分析策略,如是家族性,遗传方式为 A D,可考虑行 A CTN4、TRPC6和INF2分析;遗传方式为AR,可行NPHS2分析。如果是散发性,可以不用进行基因分析,或行 N PHS2 Q229R位点分析,如果存在NPHS2 Q229R,可考虑进行全NPHS2 基因分析[12,14,16,79]。
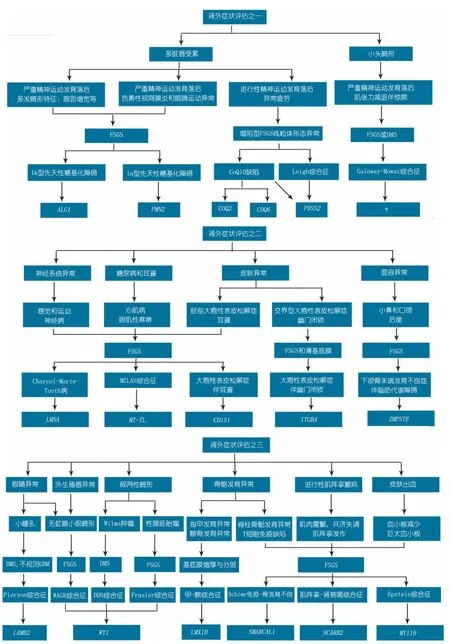
图1 综合征型SRNS基因检测流程图

图2 非综合征型SRNS基因检测流程图
5 SRNS相关致病基因临床分析的意义
SRNS 发病机制和治疗一直困扰着儿科临床医生或肾脏病工作者。SRNS致病基因不仅能明确患者的病因和发病机制,而且可以指导治疗。指导治疗体现在不必要使用糖皮质激素和免疫抑制剂,进入ESRD的SRNS患者优先考虑肾移植治疗[15,80,81]。SRNS致病基因分析是临床提供遗传咨询的依据,这种遗传咨询不仅可以提供给患者的父母,同时也可提供给患者本人[8,77]。如父母是NPHS1突变的携带者,其子女发生CNS的概率为25%,母亲在妊娠20周后应该检测α甲胎蛋白(AFP),AFP显著增高提示CNS可能性极大,必要时可行产前诊断;又如 W T1突变的患者,理论上有发生Wilm's肿瘤和肾胚胎瘤的可能,应该定期随访肾脏超声[11,37,77]。
总之,SRNS致病基因多种,SRNS患者的表型和基因型复杂。临床工作者应该首先在不同遗传背景人群构建SRNS基因突变图谱,根据图谱再构建基因分析的方案,然后依据方案选择经济、快速和准确的方法来分析SRNS致病基因。虽然基于芯片技术的多基因外显子捕获和高通量测序将是未来SRNS相关致病基因全面分析的方向,这些技术已经不是问题,但是庞大的测序数据分析将成为这项技术开展的新问题。集数学、生物学和计算机学为一体的生物信息学发展会帮助解决这些新问题。
[1]Greenbaum LA, Benndorf R, Smoyer WE.Childhood nephrotic syndrome-current and future therapies.Nat Rev Nephrol,2012,8(8):445-458
[2]Davin JC, Rutjes NW.Nephrotic syndrome in children:from bench to treatment.Int J Nephrol,2011,372304
[3]Machado JR, Rocha LP, Neves PD, et al.An overview of molecular mechanism of nephrotic syndrome.Int J Nephrol,2012,937623
[4] Patrakka J, Tryggvason K.New insights into the role of podocytes in proteinuria.Nat Rev Nephrol,2009,5(8):463-468
[5]Lennon R, Watson L, Webb NJ.Nephrotic syndrome in children.Paediatr and child health,2012,20(1):36-42
[6]Sinha A, Bagga A.Nephrotic syndrome.Indian J Pediatr,2012,79(8):1045-1055
[7]Saleem MA.New developments in steroid-resistant nephrotic syndrome.Pediatr Nephrol, 2013, 28(5):699-709
[8]Benoit G, Machuca E, Antignac C.Hereditary nephrotic syndrome:a systematic approach for genetic testing and a review of associated podocyte gene mutations.Pediatr Nephrol,2010,25(9):1621-1632
[9]Mccarthy HJ, Saleem MA.Genetics in clinical practice:nephrotic and proteinuric syndromes.Nephron Exp Nephrol,2011,118(1):1-8
[10]Liapis H.Molecular pathology of nephrotic syndrome in childhood:a contemporary approach to diagnosis.Pediatr Dev Pathol,2008,11(4):154-163
[11]Lowik MM, Groenen PJ, Levtchenko EN, et al.Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis-a review.Eur J Pediatr,2009,168(11):1291-1304
[12]D'Agati VD, Kaskel FJ, Falk RJ.focal segmental glomerulosclerosis.N Engl J Med,2011, 365(25):2398-2411
[13]Jungraithmayr TC, Hofer K, Cochat P, et al.Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation.J Am Soc Nephrol,2011,22(3):579-585
[14]Santin S, Bullich G, Tazon-Vega B, et al.Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome.Clin J Am Soc Nephrol,2011,6(5):1139-1148
[15]Gbadegesin RA, Winn MP, Smoyer WE.Genetic testing in nephrotic syndrome-challenges and opportunities.Nat Rev Nephrol,2013,9(3):179-184
[16]Lipska BS, Iatropoulos P, Maranta R, et al.Genetic screening in adolescents with steroid-resistant nephrotic syndrome.Kidney Int,2013, 84(1):206-213
[17]Gubler MC.Nephrotic syndrome:Genetic testing in steroidresistant nephrotic syndrome.Nat Rev Nephrol,2011,7(8):430-431
[18]The Subspecialty Group of Nephrology, Society of Pediatrics,Chinese Medical Association(中华医学会儿科分会肾脏病学组).Evidence-based guidelines on diagnosis and treatment of childhood common renal diseases(Ⅰ)Evidence-bused guideline on diagnosis and treatment of steroid-sensitive,relapsing/steroid-dependent nephrotic syndrome(for trial implementation)Chin J Pediatr(中华儿科杂志),2009,47(3):167-169
[19]Lombel RM, Gipson DS, Hodson EM.Treatment of steroidsensitive nephrotic syndrome:new guidelines from KDIGO.Pediatr Nephrol,2013,28(3):415-426
[20]Piscione TD, Licht C.Genetics of proteinuria:an overview of gene mutations associated with nonsyndromic proteinuric glomerulopathies.Adv Chronic Kidney Dis,2011,18(4):273-289
[21]Kestila M,Lenkkeri U,Mannikko M,et al.Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome.Mol Cell,1998,1(4):575-582
[22]Lenkkeri U, Mannikko M, Mccready P, et al.Structure of the gene for congenital nephrotic syndrome of the finnish type(NPHS1)and characterization of mutations.Am J Hum Genet,1999,64(1):51-61
[23]Boute N, Gribouval O, Roselli S, et al.NPHS2, encoding the glomerular protein podocin,is mutated in autosomal recessive steroid-resistant nephrotic syndrome.Nat Genet,2000,24(4):349-354
[24]Hinkes B, Wiggins RC, Gbadegesin R, et al.Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible.Nat Genet,2006,38(12):1397-1405
[25]Ozaltin F, Ibsirlioglu T, Taskiran EZ, et al.Disruption of PTPRO causes childhood-onset nephrotic syndrome.Am J Hum Genet,2011,89(1):139-147
[26]Kaplan JM, Kim SH, North KN, et al.Mutations in ACTN4,encoding alpha-actinin-4,cause familial focal segmental glomerulosclerosis.Nat Genet,2000,24(3):251-256
[27]Winn MP, Conlon PJ, Lynn KL, et al.A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis.Science,2005,308(5729):1801-1804
[28]Gigante M, Pontrelli P, Montemurno E, et al.CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis(FSGS).Nephrol Dial Transplant,2009,24(6):1858-1864
[29]Genovese G, Friedman DJ, Ross MD, et al.Association of trypanolytic ApoL1 variants with kidney disease in African Americans.Science,2010,329(5993):841-845
[30]Brown EJ, Schlondorff JS, Becker DJ, et al.Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis.Nat Genet,2010,42(1):72-76
[31]Sanna-Cherchi S, Burgess KE, Nees SN, et al.Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome.Kidney Int,2011,80(4):389-396
[32]Ozaltin F, Li B, Rauhauser A, et al.DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN.J Am Soc Nephrol,2013,24(3):377-384
[33]Gupta I R, Baldwin C, Auguste D, et al.ARHGDIA:a novel gene implicated in nephrotic syndrome.J Med Genet,2013,50(5):330-338
[34]Li GM(李国民),Gao XW,Xu H, et al.Clinical characteristics and INF2 gene mutation analysis in a family with autosomal dominant focal segmental glomerulosclerosis.Chin J Evid Based Pediatr(中国循证儿科杂志),2013,8(3):192-196
[35]Genovese G, Tonna SJ, Knob AU, et al.A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9.Kidney Int,2010,78(7):698-704
[36]Rosset S, Tzur S, Behar DM, et al.The population genetics of chronic kidney disease:insights from the MYH9-APOL1 locus.Nat Rev Nephrol,2011,7(6):313-326
[37]Pollak MR.Expanding the spectrum of NPHS1-associated disease.Kidney Int,2009,76(12):1221-1223
[38]Ruf RG, Schultheiss M, Lichtenberger A, et al.Prevalence of WT1 mutations in a large cohort of patients with steroidresistant and steroid-sensitive nephrotic syndrome.Kidney Int,2004,66(2):564-570
[39]Matejas V, Al-Gazali L, Amirlak I, et al.A syndrome comprising childhood-onset glomerular kidney disease and ocular abnormalities with progressive loss of vision is caused by mutated LAMB2.Nephrol Dial Transplant,2006,21(11):3283-3286
[40]Kopp JB.Glomerular pathology in autosomal dominant MYH9 spectrum disorders:what are the clues telling us about disease mechanism?.Kidney Int,2010,78(2):130-133
[41]Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, et al.COQ2 nephropathy:a newly described inherited mitochondriopathy with primary renal involvement.J Am Soc Nephrol,2007,18(10):2773-2780
[42]Heeringa SF, Chernin G, Chaki M, et al.COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness.J Clin Invest,2011,121(5):2013-2024
[43]Kambham N, Tanji N, Seigle RL, et al.Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa.Am J Kidney Dis,2000,36(1):190-196
[44]Nicolaou N, Margadant C, Kevelam SH, et al.Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome.J Clin Invest,2012,122(12):4375-4387
[45]Elizondo LI, Cho KS, Zhang W, et al.Schimke immunoosseous dysplasia:SMARCAL1 loss-of-function and phenotypic correlation.J Med Genet,2009,46(1):49-59
[46]Berkovic SF, Dibbens LM, Oshlack A, et al.Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis.Am J Hum Genet,2008,82(3):673-684
[47]Lopez LC,Schuelke M,Quinzii CM,et al.Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2(PDSS2)mutations.Am J Hum Genet,2006,79(6):1125-1129
[48]Boyer O, Woerner S, Yang F, et al.LMX1B Mutations Cause Hereditary FSGS without Extrarenal Involvement.J Am Soc Nephrol,2013,24(8):1216-1222
[49]Agarwal AK, Zhou XJ, Hall RK, et al.Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency.J Investig Med,2006,54(4):208-213
[50]Perez-Duenas B, Garcia-Cazorla A, Pineda M, et al.Longterm evolution of eight Spanish patients with CDG type Ia:typical and atypical manifestations.Eur J Paediatr Neurol,2009,13(5):444-451
[51]Kranz C, Denecke J, Lehle L, et al.Congenital disorder of glycosylation type Ik(CDG-Ik):a defect of mannosyltransferase I.Am J Hum Genet,2004,74(3):545-551
[52]Imachi H, Murao K, Ohtsuka S, et al.A case of Dunnigantype familial partial lipodystrophy(FPLD)due to lamin A/C(LMNA)mutations complicated by end-stage renal disease.Endocrine,2009,35(1):18-21
[53]Kurogouchi F, Oguchi T, Mawatari E, et al.A case of mitochondrial cytopathy with a typical point mutation for MELAS,presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation.Am J Nephrol,1998,18(6):551-556
[54]Chernin G, Vega-Warner V, Schoeb DS, et al.Genotype/phenotype correlation in nephrotic syndrome caused by WT1 mutations.Clin J Am Soc Nephrol,2010,5(9):1655-1662
[55]Hasselbacher K, Wiggins RC, Matejas V, et al.Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders.Kidney Int,2006,70(6):1008-1012
[56]Iijima K, Someya T, Ito S, et al.Focal segmental glomerulosclerosis in patients with complete deletion of one WT1 allele.Pediatrics,2012,129(6):1621-1625
[57]Lee H, Han KH, Lee SE, et al.Urinary exosomal WT1 in childhood nephrotic syndrome.Pediatr Nephrol,2012,27(2):317-320
[58]Sartelet H, Pietrement C, Noel LH, et al.Collapsing glomerulopathy in Galloway-Mowat syndrome:a case report and review of the literature.Pathol Res Pract,2008,204(6):401-406
[59]Fletcher J, Mcdonald S, Alexander SI.Prevalence of genetic renal disease in children.Pediatr Nephrol,2013,28(2):251-256
[60]Rood IM, Deegens JK, Wetzels JF.Genetic causes of focal segmental glomerulosclerosis:implications for clinical practise.Nephrol Dial Transplant,2012, 27(3):882-890
[61]Ovunc B, Ashraf S, Vega-Warner V, et al.Mutation analysis of NPHS1 in a worldwide cohort of congenital nephrotic syndrome patients.Nephron Clin Pract,2012,120(3):139-146
[62]Santin S, Tazon-Vega B, Silva I, et al.Clinical value of NPHS2 analysis in early-and adult-onset steroid-resistant nephrotic syndrome.Clin J Am Soc Nephrol,2011,6(2):344-354
[63]Frishberg Y, Rinat C, Megged O, et al.Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children.J Am Soc Nephrol,2002,13(2):400-405
[64]Benoit G, Machuca E, Nevo F, et al.Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome.Pediatr Nephrol,2010,25(3):445-451
[65]Mottl AK, Lu M, Fine CA, et al.A novel TRPC6 mutation in a family with podocytopathy and clinical variability.BMC Nephrol,2013,14:104
[66]Boyer O, Benoit G, Gribouval O, et al.Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome.J Med Genet,2010,47(7):445-452
[67]Barua M, Brown EJ, Charoonratana VT, et al.Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis.Kidney Int,2012, 83(2):316-322
[68]Kopp J B, Winkler CA, Nelson GW.MYH9 genetic variants associated with glomerular disease:what is the role for genetic testing?.Semin Nephrol,2010,30(4):409-417
[69]Yu Z, Ding J, Huang J, et al.Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children.Nephrol Dial Transplant,2005,20(5):902-908
[70]Li J, Ding J, Zhao D, et al.WT1 gene mutations in Chinese children with early onset nephrotic syndrome.Pediatr Res,2010,68(2):155-158
[71]Mao J, Zhang Y, Du L, et al.NPHS1 and NPHS2 gene mutations in Chinese children with sporadic nephrotic syndrome.Pediatr Res,2007,61(1):117-122
[72]Knob AL.Principles of genetic testing and genetic counseling for renal clinicians.Semin Nephrol,2010,30(4):431-437
[73]Guay-Woodford LM, Knoers NV.Genetic testing:considerations for pediatric nephrologists.Semin Nephrol,2009,29(4):338-348
[74]Lee JH, Han KH, Lee H, et al.Genetic basis of congenital and infantile nephrotic syndromes.Am J Kidney Dis,2011,58(6):1042-1043
[75]Mccarthy HJ, Bierzynska A, Wherlock M, et al.Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome.Clin J Am Soc Nephrol,2013,8(4):637-648
[76]Kopp JB.An Expanding Universe of FSGS Genes and Phenotypes:LMX1B Mutations Cause Familial Autosomal Dominant FSGS Lacking Extrarenal Manifestations.J Am Soc Nephrol,2013,24(8):1183-1185
[77]Joshi S, Andersen R, Jespersen B, et al.Genetics of steroidresistant nephrotic syndrome:a review of mutation spectrum and suggested approach for genetic testing.Acta Paediatr,2013,102(9):844-856
[78]Hinkes B G, Mucha B, Vlangos CN, et al.Nephrotic syndrome in the first year of life:two thirds of cases are caused by mutations in 4 genes(NPHS1,NPHS2,WT1,and LAMB2).Pediatrics,2007,119(4):907-919
[79]Kerti A, Csohany R, Szabo A, et al.NPHS2 p.V290M mutation in late-onset steroid-resistant nephrotic syndrome.Pediatr Nephrol,2013,28(5):751-757
[80]Greenbaum LA, Benndorf R, Smoyer WE.Childhood nephrotic syndrome--current and future therapies.Nat Rev Nephrol,2012,8(8):445-458
[81]Chiang CK, Inagi R.Glomerular diseases:genetic causes and future therapeutics.Nat Rev Nephrol,2010,6(9):539-554
