DFT方法研究酸性沸石上苯与乙烯烷基化反应的机理
2013-11-21孙晓岩项曙光
张 达,孙晓岩,项曙光
(青岛科技大学 炼油化工高新技术研究所,山东 青岛 266042)
乙苯主要是用来作为苯乙烯生产中的一个中间体,苯乙烯主要用于工业上合成树脂,橡胶等,也可以用于涂料和医药产业[1]. 乙苯主要由苯和乙烯烷基化制得[2],烷基化反应所需传统催化剂大多是如AlCl3和BF3这样的有腐蚀性并且难以处理的氯化物液体催化剂[3]. 沸石催化剂的应用为乙苯的生产提供了一种环保的路线,并且通过控制沸石孔径大小,使实现沸石催化剂卓越的产品选择性成为可能[4].
大部分对酸性沸石催化苯烷基化反应的研究都集中于用分子动力学方法研究反应物和产物在沸石孔道中的扩散和吸附[5-7]. 量子化学理论方法可以从微观角度探讨酸性分子筛催化剂上的反应机理,了解反应过程中的中间产物、过渡态和主要副产物等,深化对反应机理和主要影响因素的认识,从微观角度指导催化剂的设计.
为进一步了解酸性沸石上苯烷基化反应的机理,作者使用DFT方法,以一个4T团簇代表酸性沸石,针对苯与乙烯在酸性沸石上发生烷基化反应生成乙苯的反应历程进行了系统研究,从生成能和反应活化能角度分析并讨论了苯与乙烯的反应机理.
1 计算模型和计算方法
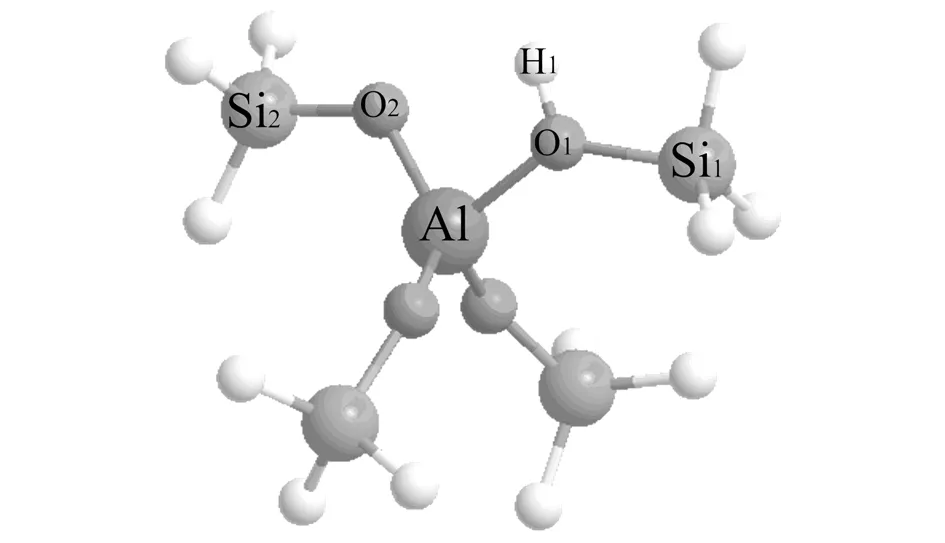
图1 分子筛4T模型Fig.1 4T cluster model of zeolite
选取了一个4T原子团簇模型来代表酸性沸石结构,如图1所示,B酸质子落在桥氧原子O1上. 模型末端H原子全部固定,以保证优化后模型结构与原沸石结构一致. 使用B3LYP密度泛函和6-31G(d)基组[8-10],进行苯与乙烯反应体系的几何优化及过渡态计算. 对于优化后的几何形状进行B3LYP/6-311G**,B3LYP/cc-pVTZ和MP2/6-311G**单点能计算. 所有的计算都用Gaussian 03程序包[10]来完成. 利用计算得到的体系能量和几何结构数据来研究分子筛上苯与乙烯的反应过程.
2 结果与讨论
2.1 反应物与产物在B酸位上的吸附

2.2 苯与乙烯烷基化反应的机理
目前,沸石上芳烃的烷基化机理还存在争议,争议的两种机理分别是Langmuir-Hinshelwood (LH)机理和Rideal-Eley (RE)机理[15]. 这里分别称其为联合机理和分步机理.
2.2.1 苯与乙烯烷基化的联合反应机理
讨论了两种不同的联合反应机理,两种机理的过渡态结构见图2. NEILS 等人[16]研究烷基化反应机理时,同样讨论了两种不同的联合反应机理. 这两种过渡态结构的主要区别在于,第一个反应过程,质子落在了原来的桥氧原子上,而第二个反应过程,质子落在了相邻的桥氧原子上. 两种反应过程的能量变化如图3所示,优化后的反应物(cR)、产物(cP)和过渡态(cTS)的几何参数列于表1. 联合反应开始于苯和乙烯在沸石酸性位上的共吸附,共吸附的吸附能为-36.92 kJ/mol,这与NIELS H 等人[16]计算苯与乙烯在H-ZSM-5分子筛上的共吸附能-34.02 kJ/mol较为接近.
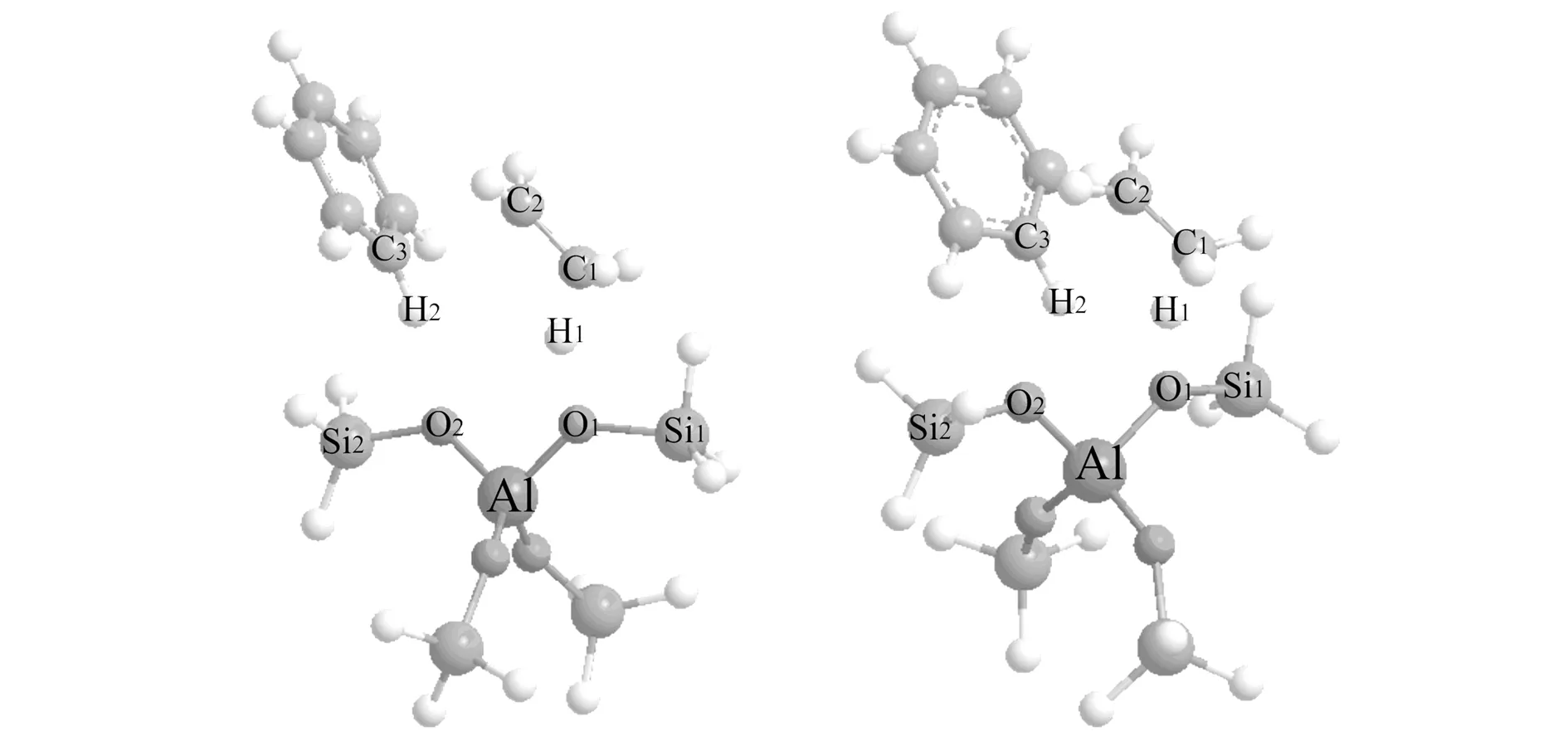
图2 联合机理的两种过渡态结构Fig.2 Optimized molecular structure of transition state for concerted mechanism


图3 两种联合反应机理的能量(kJ/mol)关系图Fig.3 Energy profile for the concerted mechanism

第二个联合机理的过渡态结构如图2(b)所示,与图2(a)所示的第一个联合机理不同处在于,酸性质子在反应后又重新回到了反应前它所在的位置. 两种机理有相似的吸附能;然而图2(b)所示的机理的活化能要低8.84 kJ/mol. 相比起第一个机理,过渡态原子键组成和键破裂之间距离更长了(即C2-C3和H1-O1/ H2-O2). NEILS等人[16]也得出类似的规律. 沿反应坐标方向的与振动有关的虚频为-222.15 i cm-1. 产物乙苯所需的解吸能为36.38 kJ/mol.

表1 联合反应机理中反应物、产物和过渡态的几何参数Table 1 Geometric parameters of reactants, products, transition states for the concerted mechanism
*键长单位nm;键角单位°
2.2.2 苯与乙烯烷基化的分步反应机理
图4所示为分步反应机理. 中间体(sE)和过渡态(sTS)的几何参数列于表2. 首先乙烯吸附在酸性位上,然后质子化的乙烯形成了乙醇盐中间物种,这步反应的活化能为122.47 kJ/mol. 这与NIELS HANSEN等人[16-17]的结果119.7和126.25 kJ/mol比较接近. 过渡态(sTS(a))中O1-H1键的键长从0.098 4 nm增加到0.134 4 nm,C1-C2键长由0.133 7 nm增加到0.139 7 nm. 过渡态相关的虚频为-868.32 i cm-1. 乙醇盐中间物种比以π-π键吸附于酸性位的乙烯更加稳定64.13 kJ/mol. 第二步反应中,乙醇盐中间物种与苯之间存在弱相互作用,并与扩散到附近的苯分子发生反应,乙基与苯上的碳原子成键,同时苯上的一个质子还给沸石桥氧原子. 在反应过程中,乙醇盐中间物种的C2-O2共价键在乙基与苯的成键和苯上的质子向沸石框架移动时断裂. 计算得此反应过程的活化能为190.24 kJ/mol,比联合反应机理要高约30 kJ/mol. 有研究显示,乙醇盐中间物种的稳定性,很大程度上受到沸石孔道壁空间位阻的影响[18]. 当模型的几何结构具有沸石的结构特性时,会削弱乙醇盐中间物种与沸石间的共价键,并且由于分子筛孔道壁空间位阻的约束大大降低了乙醇盐中间物种的稳定性[18-19]. 过渡态中成键原子之间的距离(C2和C3)明显短于(短了0.04~0.05 nm)联合反应机理. 过渡态沿反应坐标方向的振动模式的虚频为201.85 i cm-1.
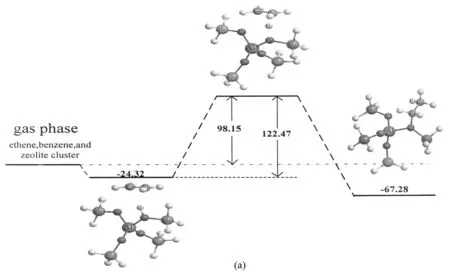

图4 分步反应机理的能量(kJ/mol)关系图Fig.4 Energy profile for the stepwise mechanism

sTS(a)sEsTS(b)Al-O10.182 30.189 60.178 0Al-O20.180 30.171 00.177 7Si1-O10.167 50.177 00.163 9Si2-O20.166 40.163 30.163 3C1-C20.139 70.151 80.149 9C2-C30.231 3O2-C20.265 30.148 90.276 9C1-H10.129 60.110 0C2-H10.210 7C3-H20.119 0O1-H10.134 4O2-H20.146 5∠Si1O1Al132.13109.15135.32∠Si2O2Al126.37164.58135.61∠C1C2C3H2-82.55
*键长单位nm;键角单位°
由上述计算结果可知,两个联合机理的活化能分别为155.06 kJ/mol和168.08 kJ/mol. 对于分步机理,乙醇盐中间物种形成过程的活化能为122.47 kJ/mol,表观反应的活化能为190.24 kJ/mol,该步骤是速率控制步骤. 联合反应机理的活化能介于分步机理两个基元反应能垒之间. 因为联合机理的活化能较小,它占据整体烷基化反应的主导地位. 然而,从能量角度看,乙醇盐中间物种的形成相对容易,当乙醇盐中间物种形成后,稳定的乙醇盐中间物种对苯的吸附致使逆反应相对于生成乙苯的正反应来说更难发生,所以分步机理也会发生. 因此,分子筛上苯与乙烯的烷基化反应中,主要是联合反应机理,分步反应机理对其有一定程度的竞争.
3 结论
用DFT方法研究了以4T团簇代表的酸性沸石上苯与乙烯的烷基化反应机理,包括两个联合机理和一个分步机理.
联合机理中,首先发生的是反应物的共吸附过程,然后质子化的乙烯与吸附态的苯反应生成乙苯. 对于联合机理,考虑了两种反应历程,两者的区别在于质子回到分子筛上的位置不同. 苯上的质子回到原酸性位上的反应过程,其活化能为155.06 kJ/mol,而苯上的质子落到原酸性位相邻的另一桥氧原子上的反应过程,其活化能为168.08 kJ/mol.
对于分步机理,烷基化始于被吸附的乙烯质子化,并形成了具有表面活性的乙醇盐中间物种,此步反应活化能为122.47 kJ/mol. 苯的烷基化是通过苯分子与乙醇盐中间物种的相互作用产生的. 分步反应的速率控制步骤为乙基片段与苯的碳原子成键以及苯上碳原子与质子之间键的断裂. 计算得此过程活化能为190.24 kJ/mol. 基于对苯与乙烯反应能量的分析,可知苯与乙烯烷基化的反应机理中,联合反应机理的活化能更低,所以主要是联合反应机理. 但是由于分步反应机理中乙醇盐中间物种的形成比较容易,所以两种反应机理存在一定程度的竞争.
参考文献:
[1] DEGNAN T F,MORRIS SMITH C,VENKAT C R.Alkylation of aromatics with ethylene and propylene: recent developments in commercial processes[J].Appl Catal A,2001,221:283-294.
[2] SHI Y F,GAO Y,YUAN W K.Benzene-ethylene alkylation in near-critical regions[J].Ind Eng Chem Res,2001,40:4253-4257.
[3] BELLUSSI G,PAZZUCONI G,PEREGO C,et al.Liquid-phase alkylation of benzene with light olefins catalyzed byβ-zeolites[J].J Catal,1995,157:227-234.
[4] CHEN N Y,GARWOOD W E.Industrial application of shape-selective catalysis[J].Catal Rev Sci Eng, 1986,28:185-264.
[5] FRANK B,DAHLKE K,EMIG G,et al.A monte-carlo simulation of diffusion and reaction in zeolites[J].Microporous Materials,1993,1:43-56.
[6] SNURR R Q,BELL A T,THEODOROU D N.Prediction of adsorption of aromatic hydrocarbons in silicalite from grand canonical monte carlo simulations with biased insertions[J].J Phys Chem,1993,97:13742-13752.
[7] DEKA R C,VETRIVEL R.Computer-aided search for shape-selective zeolite catalysts for the synthesis ofp-isobutylethylbenzene[J].Chem Commun,1996,21:2397-2398.
[8] BELYAEVA V V,FROLOV Y L,VORONKOV M G.Dft(b3lyp) calculations of silatranes and their radical cations first ionization potentials[J].J Struc Chem,2005,46:1072-1076.
[9] HAYASE S,HROVAT D A,BORDEN W T.Ab3lyp study of the effects of phenyl substituents on 1,5-hydrogen shifts in 3-(z)-1,3-pentadiene provides evidence against a chameleonic transition structure[J].J Am Chem Soc,2004,126:10028-10034.
[10] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 03, Revision C02[CP]. Gaussian Inc., Pittsburgh, PA., 2003.
[11] SIFFERT S L,GAILLARD B,SU L.Alkylation of benzene by propene on a series of beta zeolites:toward a better understanding of the mechanisms[J].J Mol Catal A,2000, 153: 267-279.
[12] YINGCHUN DU,HAI WANG,SHU CHEN.Study on alkylation of benzene with ethylene overβ-zeolite catalyst to ethylbenzene by in situ IR[J].J Mol Catal A,2002,179:253-261.
[13] VENUTO P B,LANDIS P S.Organic catalysis over crystalline aluminosilicates[J].Adv Catal,1968, 18:259-371.
[14] FLEGO C,KIRICSI I,PEREGO C,et al.Adsorption of propene,benzene,their mixtures and cumene on h-beta zeolites studied by IR and UV-Vis spectroscopy[J].Stud Surf Sci Catal,1995,94:405-412.
[15] SMIRNIOTIS P G, RUCKENSTEIN E. Alkylation of benzene or toluene with meoh or chover zsm-5 or2zeolite: effect of the zeolite pore openings and of the hydrocarbons involved on the mechanism of alkylation [J].Ind Eng Chem Res,1995,34:1517-1528.
[16] NIELS HANSEN,TILL BRUÜGGEMANN,ALEXIS T BELL,et al.Theoretical investigation of benzene alkylation with ethene over H-zsm-5[J].J Phy Chem C,2008,112:15402-154011.
[17] NAMUANGRUK S,PANTU P,LIMTRAKUL J.Alkylation of benzene with ethylene over faujasite zeolite investigated by the oniom method[J].J Catal,2004,225:523-530.
[18] BORONAT M,ZIRCOVICH-WILSON C M,PEDRO VIRUELA,et al.Influence of the local geometry of zeolite active sites and olefin size on the stability of alkoxide intermediates[J].J Phys Chem B,2001,105:11169-11177.
[19] ROZANSKA X,DEMUTH T H,HUTSCHKA F,et al.A periodic structure density functional theory study of propylene chemisorption in acidic chabazite:effect of zeolite structure relaxation[J].J Phys Chem B,2002,106:3248-3254.
