氯化锆催化芳胺和正丁醛合成喹啉衍生物
2013-11-21郭巧霞饶莎莎任申勇
郭巧霞,饶莎莎,任申勇,徐 佳
(中国石油大学(北京) 理学院,北京 102249)
喹啉衍生物是一类重要的含氮杂环化合物,其主要骨架喹啉广泛存在于骨油和煤焦油中. 喹啉及其衍生物在医药、农药、染料以及化学助剂等领域有着重要的应用[1-4]. 随着这类化合物在各领域的广泛应用,该类化合物的合成已成为国内外的研究热点. 喹啉可以从煤焦油中提取,但是,喹啉类衍生物却要用化学合成的方法制备. 近年来,人们对于新型喹啉衍生物的合成、新合成路线和新型催化剂体系的研究越来越活跃. 其制备方法包括传统合成方法和现代合成方法两种. 传统的合成方法主要有Skraup合成法[5]、Doebner-Von Miller合成法[6]、Friedländer合成法[7]]、Camps合成法[8]以及周环反应合成法[9];现代合成方法有微波辐射促进合成法[10]、有机金属催化合成法[11]、高氯酸负载二氧化硅催化合成法[12]等. 在这些合成方法中,Doebner-Von Miller合成法由于原料易得、操作简单、反应条件温和而受到人们的广泛关注. 但是很多反应存在制备条件苛刻、原料和催化剂价格昂贵、对设备要求高、对环境造成污染等问题,不适于工业化生产. 近几年科学家们一直致力于研究Doebner-Von Miller合成法的催化剂. WANG等[13]用单质碘取代原来的浓盐酸或者浓硫酸作催化剂制得了喹啉衍生物. 本课题组分别以对甲苯磺酸[14]和AlCl3[15]为促进剂,研究了喹啉衍生物的合成反应,对反应促进剂、反应条件、原料配比以及氧化剂H2O2的促进作用等进行了考察. 当用对甲基苯磺酸(TsOH)为促进剂时,3-乙基-2-丙基喹啉收率可达到59%,同时生成40%的N-丁基苯胺. 当以AlCl3为促进剂,H2O2为氧化剂时,喹啉收率由64%提高到84%,生成喹啉衍生物的选择性达100%. 虽然用TsOH或AlCl3作促进剂时,试剂的价格便宜,喹啉的收率也高,但是前者是计量反应,所用促进剂的用量较大,兼之两种促进剂均不能重复使用,造成原料成本高. 此外,反应后处理需要用碱淬灭反应,产生了很多环境问题. 因此寻找新的催化体系,降低催化剂的用量和价格,依然是我们的研究重点. 本文作者以ZrCl4为催化剂,考察了空气、催化剂用量、原料比例、不同的反应底物(醛及芳胺上取代基)对反应的影响(图1),探索了一条工艺条件稳定、操作简便、价格便宜的合成方法,并对所合成的一系列喹啉衍生物进行了高分辨质谱和核磁共振光谱表征.
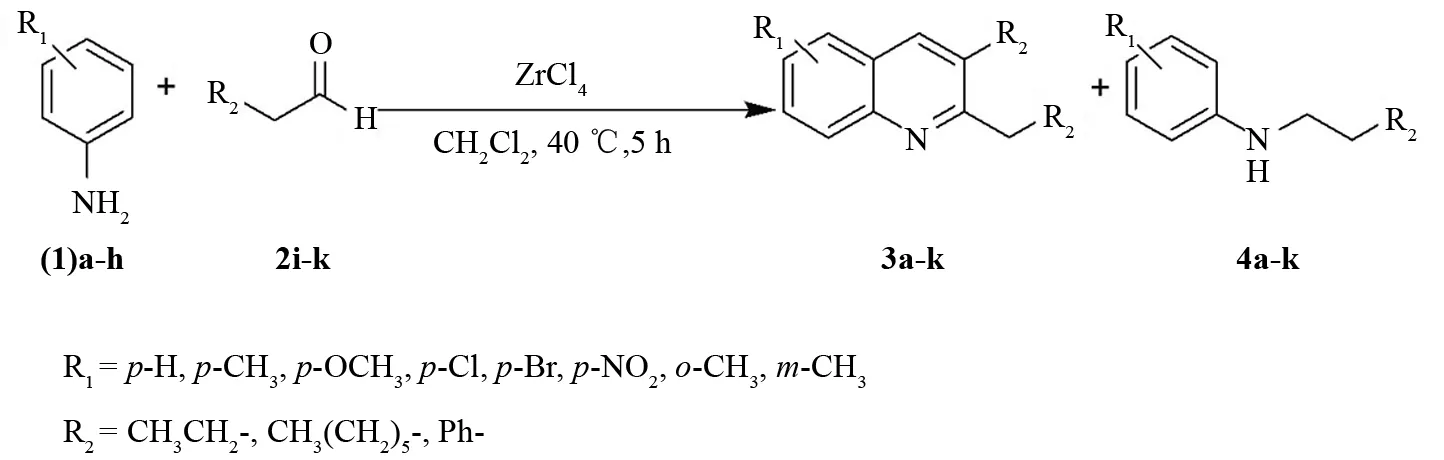
图1 不同芳胺和醛的反应Fig.1 The reaction of different aldehydes and arylamine
1 实验部分
1.1 原料及仪器
芳胺、正丁醛、二氯甲烷、正十二烷、无水四氯化锆等原料均为市售分析纯试剂,没有进一步纯化处理;反应监测采用GC-14B型气相色谱仪(日本岛津公司),产物鉴定采用JNM-LA300型核磁共振仪(日本电子株式会社).
1.2 实验方法
取干燥洁净的50 mL Schlenk管,按照一定的比例向Schlenk管内依次加入0.111 5 g(0.5 mmol)的ZrCl4、50 mL二氯甲烷,溶液呈白色浑浊,再加入0.45 mL(5 mmol)苯胺,溶液变成黄色浑浊,然后加入1.8 mL(20 mmol)正丁醛,溶液变成黄绿色,加热并保持反应在40 ℃下进行,用气相色谱仪和TLC检测反应的进程. 反应结束后,用3 mol/L氨水淬灭反应,再用3×20 mL的乙醚萃取有机相,经旋转蒸发仪浓缩后得到黄色黏稠液体,用硅胶柱分离提纯产品,洗脱剂为体积比为10∶1的石油醚/乙醚混合液. 得到0.53 g 3-乙基-2-丙基喹啉(3a)的黄色黏稠液,分离收率为53%,同时得到0.12 g副产物N-丁基苯胺(4a),分离收率为16%.3a核磁共振表征数据为:1H NMR(CDCl3, Me4Si)δ: 1.07(t,J= 7.2 Hz, 3H), 1.35(t,J= 7.5 Hz, 3H), 1.76~1.89 (m, 2H), 2.84(q,J= 7.5 Hz, 2H), 2.96(t,J= 8.1 Hz, 2H), 7.45(t,J= 7.2 Hz, 1H), 7.62(t,J= 7.2 Hz, 1H), 7.73(d,J= 8.1 Hz, 1H), 7.87(s, 1H), 8.02(d,J= 8.4 Hz, 1H).13C NMR(CDCl3, Me4Si)δ: 14.30, 14.38, 22.84, 25.12, 37.67, 125.52, 126.85, 127.31, 128.31, 128.35, 133.90, 135.34, 146.32, 161.96. C14H17N+Na分子量计算值:222.125 9,高分辨质谱测定值:222.125 4.
6-甲基-3-乙基-2-丙基喹啉(3b):以对甲苯胺和正丁醛为原料,上述相同条件下反应8 h,色谱收率为65%,分离收率为60%,核磁表征数据为:1H NMR (CDCl3, Me4Si)δ: 1.07(t,J= 7.4 Hz, 3H), 1.33(t,J= 7.6 Hz, 3H), 1.78~1.85(m, 2H), 2.50(s, 3H), 2.82(q,J= 7.6 Hz, 2H), 2.94(t,J= 7.9 Hz, 2H) , 7.44(d,J= 8.3 Hz, 1H) , 7.48(s, 1H) , 7.76(s, 1H), 7.90(d,J= 8.2 Hz, 1H).13C NMR (CDCl3, Me4Si)δ: 14.5, 14.6, 21.6, 23.0, 25.3, 37.8, 125.9, 127.3, 128.2, 130.6, 133.4, 135.3, 135.4, 145.1, 161.1. C15H19N+Na分子量计算值:236.141 5,高分辨质谱测定值:236.140 8.
4-甲基-N-丁基苯胺(4b):色谱收率为28%,分离收率为21%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.95(t,J= 7.2 Hz, 3H), 1.39~1.45(m, 2H), 1.56~1.61(m, 2H), 2.23(s, 3H), 3.08(t,J= 7.2 Hz, 2H), 3.48(s, 1H), 6.53(d,J= 8.4 Hz, 2H), 6.97(d,J= 7.8 Hz, 2H).13C NMR(CDCl3, Me4Si)δ: 14.02, 20.42, 20.46, 31.87, 44.18, 113.02, 126.37, 129.80, 146.44.
6-甲氧基-3-乙基-2丙基喹啉(3c):以对甲氧基苯胺和正丁醛为原料,上述相同条件下反应6 h,色谱收率为65%,分离收率为59%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 1.06(t,J= 7.2 Hz, 3H), 1.33(t,J= 7.4 Hz, 3H), 1.79~1.84(m, 2H), 2.81(q, 4J= 7.5 Hz, 2H), 2.91(t,J= 8.0 Hz, 2H), 3.91(s, 3H), 7.01(s, 1H), 7.25~7.28(m, 1H), 7.76(s, 1H), 7.90(d,J= 9.2 Hz, 1H).13C NMR(CDCl3, Me4Si)δ: 14.4, 14.6, 23.0, 25.2, 37.7, 55.6, 104.7, 120.8, 128.2, 130.0, 133.0, 135.7, 142.6, 157.2, 159.5. C15H19NO+Na分子量计算值:252.136 4,高分辨质谱测定值:252.135.
4-甲氧基-N-丁基苯胺(4c):色谱收率为15%,分离收率为8%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.95(t,J= 7.2 Hz, 3H), 1.39~1.45(m, 2H), 1.56~1.60(m, 2H), 3.06(t,J= 6.9 Hz, 2H), 3.73(s, 3H), 3.81(s, 1H), 6.57(d,J= 8.4 Hz, 2H), 6.77(d,J= 9.0 Hz, 2H).13C NMR(CDCl3, Me4Si)δ: 14.00, 20.40, 31.90, 44.80, 55.93, 114.11, 115.02, 142.99, 152.07.
6-氯-3-乙基-2-丙基喹啉(3d):以对氯苯胺和正丁醛为原料,上述相同条件下反应6 h,色谱收率为56%,分离收率为46%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 1.06(t,J= 7.4 Hz, 3H), 1.33(t,J= 7.6 Hz, 3H), 1.80~1.86(m, 2H), 2.82(q,J= 7.5 Hz, 2H), 2.94(t,J= 7.8 Hz, 2H), 7.54(d,J= 8.8 Hz, 1H), 7.70(s, 1H), 7.76(s, 1H), 7.93(d,J= 8.8 Hz, 1H).13C NMR(CDCl3, Me4Si)δ: 14.3, 14.4, 22.8, 25.2, 37.8, 125.7 128.0, 129.2, 130.2, 131.2, 132.9, 136.5, 144.8, 162.4. C14H16NCl分子量计算值:234.105 0,高分辨质谱测定值:234.104 4.
4-氯-N-丁基苯胺(4d):色谱收率为24%,分离收率为20%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.95(t,J= 7.2 Hz, 3H), 1.39~1.45(m, 2H), 1.56~1.61(m, 2H), 3.07(t,J= 7.2 Hz, 2H), 3.63(s,1H), 6.51(d,J= 8.4 Hz,2H), 7.1(d,J= 9.0 Hz, 2H).13C NMR(CDCl3, Me4Si)δ: 13.94, 20.33, 31.61, 43.89, 113.79, 121.67, 129.08, 147.14. 6-溴-3-乙基-2-丙基喹啉(3e):以对溴苯胺和正丁醛为原料,上述相同条件下反应6 h,色谱收率为58%,分离收率为55%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 1.06(t,J= 7.4 Hz, 3H), 1.33(t,J= 7.4 Hz, 3H), 1.80~1.86(m, 2H), 2.82(q,J= 7.5 Hz, 2H), 2.92(t,J= 7.8 Hz, 2H), 7.64~7.67(m, 1H), 7.74(s, 1H), 7.85-7.87(m, 2H).13C NMR(CDCl3, Me4Si)δ: 14.3, 14.4, 22.7, 25.2, 37.8, 119.3, 128.6, 129.0, 130.4, 131.7, 132.8, 136.5, 145.0, 162.6. C14H16NBr分子量计算值:278.054 4,高分辨质谱测定值:278.053 9.
4-溴-N-丁基苯胺(4e):色谱收率为19%,分离收率为11%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.95(t,J= 7.2 Hz, 3H), 1.39~1.45(m, 2H), 1.56~1.61(m, 2H), 3.06(t,J= 7.2 Hz, 2H), 3.61(s, 1H), 6.46(d,J= 9.0 Hz, 2H), 7.23(d,J= 8.4 Hz, 2H).13C NMR(CDCl3, Me4Si)δ: 13.95, 20.34, 31.59, 43.76, 108.60, 114.28, 131.95, 147.58. 6-硝基-3-乙基-2-丙基喹啉(3f):以对硝基苯胺和正丁醛为原料,上述相同条件下反应8 h,色谱收率为15%,分离收率为11%,核磁表征数据为:1H NMR (CDCl3, Me4Si)δ: 1.09(t,J= 7.4 Hz, 3H), 1.38(t,J= 7.5 Hz, 3H), 1.82~1.94(m, 2H), 2.89(q,J= 7.6 Hz, 2H), 3.00(t,J= 7.9 Hz, 2H), 8.02(s, 1H), 8.11(d,J= 9.3 Hz, 1H), 8.37(d,J= 12.4 Hz, 1H), 8.71(s, 1H).13C NMR (CDCl3, Me4Si)δ: 14.1, 14.4, 22.4, 25.2, 38.0, 121.9, 123.9, 126.1, 130.3, 135.1, 137.8, 145.0, 148.6, 166.4. C14H16N2O2+Na分子量计算值:267.110 9,高分辨质谱测定值:267.110 4.
8-甲基-3-乙基-2-丙基喹啉(3g):以邻甲苯胺和正丁醛为原料,上述相同条件下反应7 h,色谱收率为67%,分离收率为55%,核磁表征数据为:1H NMR (CDCl3, Me4Si)δ: 1.12(t,J= 7.2 Hz, 3H), 1.35(t,J= 7.2 Hz, 3H), 1.95~2.01(m, 2H), 2.84(q,J= 7.8 Hz, 2H), 2.84(s, 3H), 2.99(t,J= 7.8 Hz, 2H), 7.35(t,J= 7.8Hz, 1H), 7.46(d,J= 6.6 Hz, 1H), 7.57(d,J= 8.4 Hz, 1H), 7.81(s, 1H).13C NMR (CDCl3, Me4Si)δ: 14.28, 14.35, 17.80, 21.80, 25.13, 37.42, 124.82, 125.18, 127.12, 128.21, 133.62, 135.02, 136.64, 145.42, 160.17. C15H19N+Na分子量计算值:236.141 5,高分辨质谱测定值:236.141 1.
7-甲基-3-乙基-2-丙基喹啉(3h):以间甲苯胺和正丁醛为原料,上述相同条件下反应3 h,色谱收率为66%,分离收率为55%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 1.05(t,J= 7.2 Hz, 3H), 1.31(t,J= 7.2 Hz, 3H), 1.79~1.85 (m, 2H), 2.52(s, 3H), 2.80(q,J= 7.2 Hz, 2H), 2.93(t,J= 7.8 Hz, 2H), 7.25(d,J= 8.4 Hz, 1H), 7.59(d,J= 8.4 Hz, 1H), 7.78(s, 1H), 7.79(s, 1H).13C NMR (CDCl3, Me4Si)δ: 14.42, 14.55, 21.85, 22.93, 25.21, 37.85, 125.42, 126.62, 127.67, 127.79, 133.66, 134.52, 138.42, 146.76, 161.94. C15H19N+Na分子量计算值:236.141 5,高分辨质谱测定值:236.140 8.
3-甲基-N-丁基苯胺(4h):色谱收率为37%,分离收率为22%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.95(t,J= 7.2 Hz, 3H), 1.39~1.45(m, 2H), 1.56~1.61(m, 2H), 2.26(s, 3H), 3.09(t,J= 7.2 Hz, 2H), 3.49(s, 1H), 6.40(d,J= 8.4 Hz, 1H), 6.41(s, 1H), 6.50(d,J= 7.2 Hz, 1H), 7.04(t,J= 7.8 Hz, 1H).13C NMR(CDCl3, Me4Si)δ: 14.07, 20.48, 21.78, 31.92, 43.89, 110.06, 113.66, 118.21, 129.25, 139.11, 148.78.
3-己基-2-庚基喹啉(3j):以苯胺和正辛醛为原料,上述相同条件下反应3 h,色谱收率为60%,分离收率为26%,核磁表征数据为:1H NMR(CDCl3, Me4Si)δ: 0.89~0.91(m, 6H) , 1.30~1.50(m, 14H), 1.65~1.70(m, 2H), 1.76~1.82(m, 2H), 2.77(t,J= 7.9 Hz, 2H), 2.97(t,J= 8.3 Hz, 2H), 7.42(t,J= 7.2 Hz, 1H), 7.59(t,J= 8.1 Hz, 1H), 7.69(d,J= 8.3 Hz, 1H), 7.82(s, 1H), 8.02(d,J= 8.3 Hz, 1H).13C NMR(CDCl3, Me4Si)δ: 14.2, 22.7, 22.8, 29.3, 29.4, 29.9, 30.0, 30.6, 31.8, 31.9, 32.5, 36.0, 125.6, 126.9, 127.3, 128.3, 128.6, 134.2, 134.9, 146.6, 162.4. C22H33N+Na分子量计算值:334.251 1,高分辨质谱测定值:334.250 3.
N-苯亚甲基苯胺(5):以苯胺和苯甲醛为原料,上述相同条件下反应24 h,色谱收率为94%,以水为溶剂重结晶,分离收率约为60%,核磁表征数据为:1H NMR (CDCl3, Me4Si)δ: 7.18~7.20(m, 3H), 7.34~7.35(m, 2H), 7.41~7.42(m, 3H), 7.86~7.87(m, 2H), 8.39(s, 1H).13C NMR (CDCl3, Me4Si)δ: 121.1, 126.1, 128.9, 129.0, 129.3, 131.5, 136.4, 152.3, 160.5.
2 结果与讨论
2.1 空气对反应收率的影响
空气对反应收率的影响结果如表1所示. 可以看出,不管Schlenk管是否经过无水无氧处理,3-乙基-2-丙基喹啉和N-丁基苯胺的收率变化不大,产生该现象的原因可能是在形成喹啉的过程中先生成四氢喹啉和二氢喹啉的中间体,空气中的氧气有助于这些中间体脱氢生成目标产物喹啉,由于本反应的条件比较温和,不足以使脱下的H与氧气反应生成水,因此氧气对反应的促进作用在本实验中没有表现出来. 但是,从反应结果看,氧气对反应没有抑制作用,所以为了操作方便和节约成本,后续实验都在大气环境条件下进行,即Schlenk管不再经过无水无氧处理,而是直接加料进行反应.
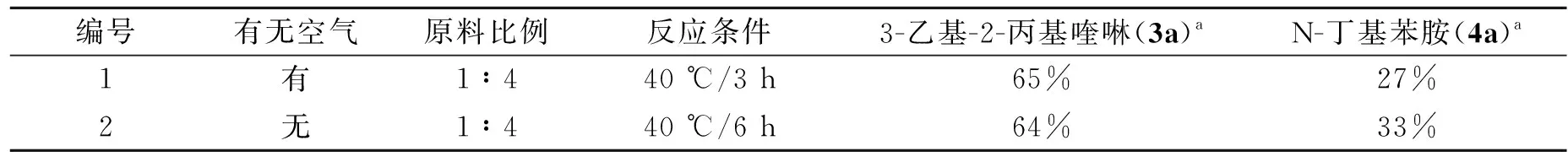
表1 空气对反应的影响Table 1 Effect of air on the reaction
注:a收率为色谱收率
2.2 溶剂对反应的影响
溶剂对反应的影响如表2所示. 实验发现,ZrCl4不溶于正己烷,并且它在正己烷中的分散度也很差,黏附在Schlenk管器壁底部,所以即使催化剂的量增加到1.5 eq,反应仍然很难在正己烷中进行. 而反应在其他几种溶剂中都能顺利的进行,虽然ZrCl4在这几种溶剂中的溶解度也不是很好,但是ZrCl4在其中的分散度却很好,能均匀分散成小颗粒而形成悬浮液,在反应的过程中能与反应物充分接触. 分别以二氯甲烷和三氯甲烷为溶剂时,3-乙基-2-丙基喹啉的收率相当,但是以二氯甲烷为溶剂生成的仲胺要比用三氯甲烷作溶剂时产生的仲胺多,且二氯甲烷价格更便宜,沸点低,后处理更容易. 故本实验确定二氯甲烷为最佳溶剂.
注:a收率为色谱收率,b以正己烷为溶剂时,即使催化剂用量增加到1.5 eq,目标产物仍然很少(微量).
2.3 催化剂用量对反应的影响
由表3可以看到,无论ZrCl4的量为0.1 eq还是1.0 eq时,反应都能顺利的进行,而且产物3-乙基-2-丙基喹啉和N-丁基苯胺的收率基本一样,可以判断该反应为催化反应. 因此,在后续的实验中,ZrCl4的量都采用为0.1 eq进行实验.

表3 ZrCl4用量对反应的影响Table 3 Effect of ZrCl4 dosage on reaction
注:a收率为色谱收率
2.4 原料摩尔比对反应的影响
由表4可以看到,当胺与醛的摩尔比为1∶1时,只有45%的3-乙基-2-丙基喹啉生成,而没有N-丁基苯胺生成. 可能是正丁醛的用量太少,其用量只够生成亚胺. 当原料苯胺和正丁醛的比例为1∶2和1∶3时,生成的3-乙基-2-丙基喹啉和N-丁基苯胺几乎不变. 当原料比例上升到1∶4时,3-乙基-2-丙基喹啉和N-丁基苯胺的收率均达到最大. 故后续实验苯胺与正丁醛的摩尔比为1∶4.
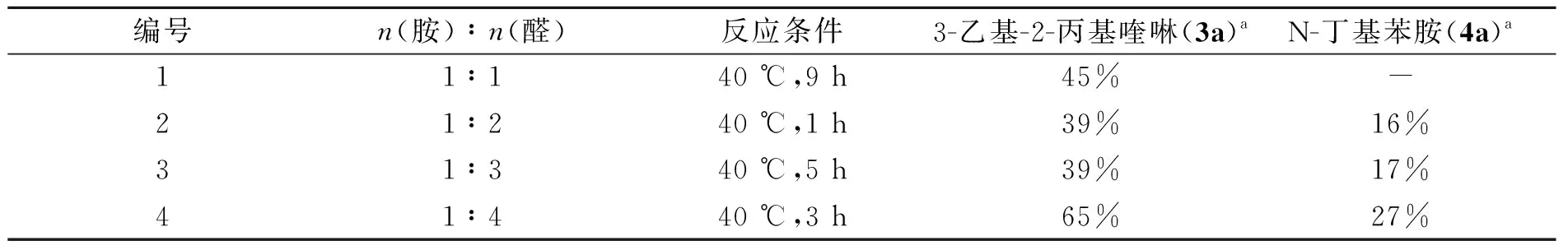
表4 原料摩尔比对反应的影响Table 4 Effect of molar ratio of starting material on the reaction
注:a收率为色谱收率
2.5 不同底物对反应的影响
实验还考察了含不同取代基的苯胺和不同种类的醛对反应的影响,其结果如表5所示. 由表5可以看出,反应底物芳胺上不管是含有供电子取代基,还是吸电子取代基,取代基不管是在氨基的对位、间位还是邻位,反应都能顺利地进行,生成喹啉衍生物和仲胺,说明本方法的适用范围比较广. 芳胺上的取代基效应比较明显,当芳胺上含有供电子取代基时产物的收率较高,当芳胺上含有吸电子取代基时收率会降低,说明供电子基有利于芳胺与醛的亲核加成消除反应,有利于中间体亚胺的生成. 而当用正辛醛为反应底物时,喹啉色谱收率可达60%,仲胺色谱收率为35%,但是它们的分离收率都很低,原因主要是正辛醛与产物很难通过硅胶柱实现分离,需要多次过柱子才能得到少量纯净的目标产物(表5中例9). 当苯胺与苯甲醛反应时,只有N-苯亚甲基苯胺5生成,色谱收率94%,用水重结晶,分离收率约60%(表5中例10). 而苯胺与苯乙醛反应生成46%的喹啉衍生物(表5中例11).
所探索的合成喹啉的方法与有关文献报道的[6,16]合成相同喹啉衍生物的方法相比,所用的催化剂价格便宜、反应条件温和、反应时间短、原料易得、收率较高且对环境的污染较少. 相对于本课题组以前的工作[14]来说,本实验所用的溶剂在以前的基础上有所优化,避免了使用毒性较大的乙腈溶剂. 本方法无需在氮气的保护下进行,使操作更加方便;反应为催化反应,催化剂用量少,目标产物2, 3-二取代喹啉衍生物的收率较高.
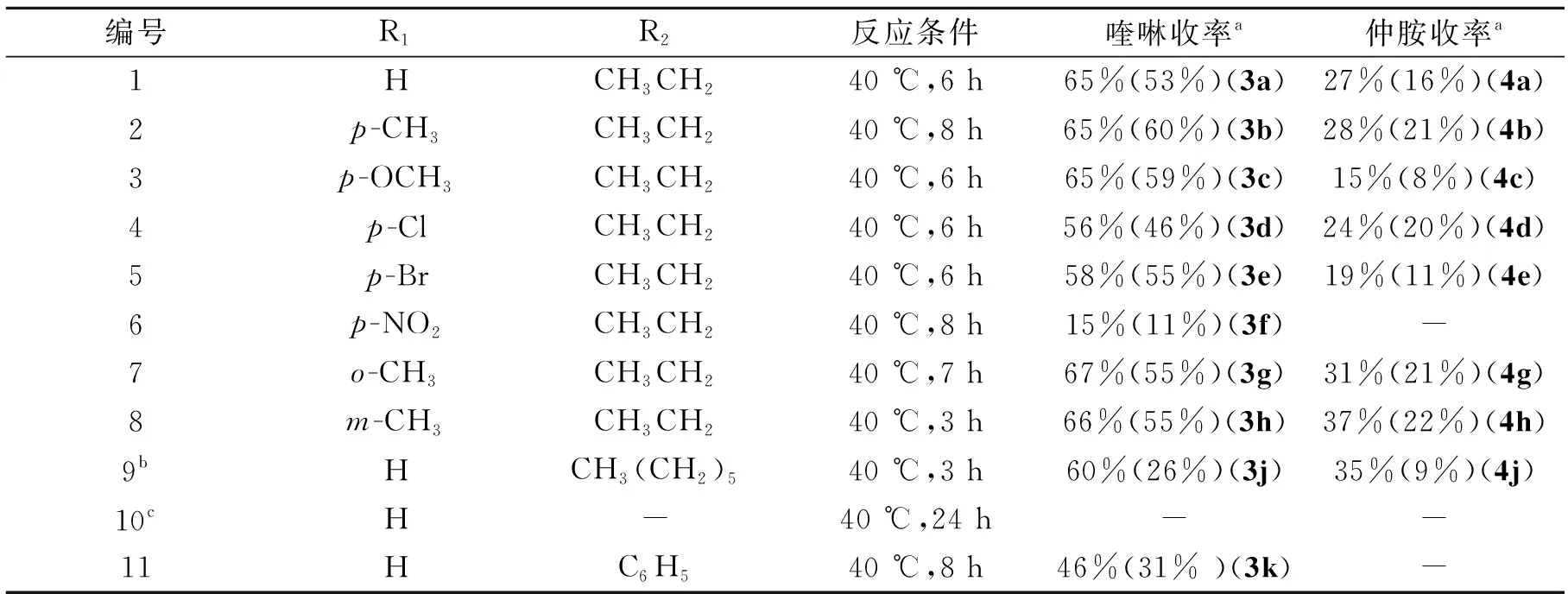
表5 不同苯胺和醛对反应的影响Table 5 Effect of different aldehydes and anilines on reaction
注:a收率为色谱收率,括号内为分离收率;b目标产物和正辛醛很难分离,经过蒸馏和四次过柱子才得到少量的纯净目标产物.
c反应底物为苯胺与苯甲醛,只有N-苯亚甲基苯胺5生成,无喹啉衍生物和仲胺生成.
虽然Doebner-Von Miller反应的机理现在还不十分清楚,但是人们可以肯定的是它与Skraup反应的机理是不同的. 通过调研相关的资料,推测生成喹啉的机理与本课题组研究的以对甲苯磺酸和三氯化铝为促进剂合成喹啉衍生物的机理类似[14,15],即苯胺与正丁醛首先发生亲核加成消除反应生成亚胺,ZrCl4的存在促进了含α氢的醛烯醇化,该烯醇式中间体再与亚胺发生Michael加成反应,环化缩合得到四氢喹啉衍生物,脱水脱氢后芳构化生成喹啉衍生物,亚胺接受氢被还原成仲胺,由于苯甲醛不含α氢原子,不能重排成烯醇式,故只生成亚胺.
3 结论
作者发展了一种新的ZrCl4催化反应体系,该体系是以苯胺衍生物和脂肪醛或含α氢的芳香醛为原料,以ZrCl4为催化剂进行反应,考察了空气、溶剂、催化剂用量以及原料比例对形成喹啉衍生物反应的影响. 结果显示,反应可在大气环境下进行,反应体系的最佳溶剂是二氯甲烷,最佳催化剂用量是0.1 eq,最佳原料比例为n(芳胺)∶n(正丁醛) = 1∶4. 在最佳反应条件下,3-乙基-2-丙基喹啉的收率可达65%,同时生成27%的仲胺. 另外,当芳胺上含有供电子取代基时有利于反应的进行,喹啉及仲胺的收率较高;而当芳胺上含有吸电子取代基时不利于反应的进行,产物的收率降低. 正辛醛作为反应底物时,产物与原料较难分离,分离收率较低. 总之,该工艺具有反应速度快、条件温和、操作简单、价格低廉以及收率高等优点.
参考文献:
[1] 徐 峰, 杨定乔, 李文辉, 等. 喹啉类药物研究进展[J]. 广东药学, 2004, 14(6): 6-9.
[2] 江上浩. 新型植物细菌病防治剂-Starna[J]. 农药译丛, 1993, 15(5): 55-61.
[3] 史学松, 徐晓友, 胡家振,等. 四氢喹啉型杂环分散染料的研究[J]. 染料工业, 1992, 29(1): 1-4.
[4] 阮传民, 徐其亨. 四个8-氨基喹啉新衍生物的合成及其分析应用[J]. 云南化工, 1991, 1(2): 61-63.
[5] CLARKE H T. DAVIS A W. Quinoline [J]. Org Synth, 1922, 2: 79-82.
[6] MATSUGI M, TABUSA F, MINAMIKAWA J. Doebner-Miller synthesis in a two-phase system: practical preparation of quinolines [J]. Tetrah Lett, 2000, 41: 8523-8525.
[7] CHENG C C, YAN S J. The Friedländer synthesis of quinolines [J]. Org React, 1982, 28: 37-201.
[8] 朱淬砺.药物合成化学[M].北京: 化学工业出版社,1999.
[9] HIBINO S, SUGINO E. A facile and alternative synthesis of quinoline nucleus using thermal cyclization of 2-azahexatriene system generated from 2-alkenyl-N-acylaniline with phosphorus oxychloride [J]. Heterocycles, 1987, 26(7): 1883-1889.
[10] ZHU Hui, YANG Ri Fang, YUN Liu Hong, et al. Facile and efficient synthesis of quinolone-4-carboxylic acids under microwave irradiation [J]. Chin Chem Lett, 2010, 21: 35-38.
[11] JACOB J, CAVALIER C M, JONES W D, et al. Cobalt-catalyzed selective conversion of diallylanilines and arylimines to quinolines [J]. J Mol Catal A: Chem, 2002, (182/183): 565-570.
[12] NARASIMHULU M, REDDY T S, MAHESH K C, et al. Silica supported perchloric acid: a mild and highly efficient heterogeneous catalyst for the synthesis of poly-substituted quinolinesviaFriedländer hetero-annulation [J]. J Mol Catal A: Chem, 2007, 266: 114-117.
[13] WANG Xiang Shan, ZHOU Jie, YANG Ke, et al. Efficient method for the synthesis of 2-(3-arylbenzo[f]quinolin-2-yl)ethanol derivatives through an unusual ring-opening of THF-involved reaction [J]. Tetrah Lett, 2011, 52: 612-614.
[14] 郭巧霞,陈利维,滕卫灵. 3-乙基-2-丙基喹啉衍生物的合成工艺研究[J]. 化学研究, 2011, 22(5): 31-37.
[15] GUO Qiao Xia, WANG Wen Nian, TENG Wei Ling, et al. Oxidant effect of H2O2for the syntheses of quinoline derivativesviaone-pot reaction of aniline and aldehyde [J]. Synth Commun, 2012, 42: 2574-2584.
[16] CHO C S, KIM T K, KIM B T, et al. Ruthenium-catalyzed reductive cyclization of nitroarenes with trialkylamines leading to quinolines [J]. J Organomet Chem, 2002, 650: 65-68.
