麦芽三糖基-β-环糊精的结构*
2013-11-21于博柏玉香金征宇吴月纯汤尚文吴进菊
于博,柏玉香,金征宇,吴月纯,汤尚文,吴进菊
1(湖北文理学院化学工程与食品科学学院,湖北襄阳,441053)2(江南大学食品科学与技术国家重点实验室,江苏无锡,214122)
β-环糊精(β-CD)作为一类重要的主体化合物,在食品、医药、精细化工、分子识别、化学分析以及催化反应等领域应用广泛。其独特刚性空腔结构能够包和或部分包和客体分子,从而形成主-客体复合物,改变客体分子物理化学或生物学特性[1]。但β-CD水溶性较差而且其溶血效应和非消化道安全性制约着其应用范围[2-4]。为了增加环糊精的水溶性,降低其对生理系统的不良影响,化学或生物改性环糊精是科学可行的解决方案。通常利用化学试剂或生物酶法将特定的分子基团接枝到母体环糊精,一方面可以有效地增加其水溶性,另一方面,侧链基团的引入会降低或消除其溶血性和肾毒性。分支环糊精便是通过引入糖基来提高β-CD水溶解度的一种策略,葡萄糖基环糊精(G-β-CD)[5-6]、麦芽糖基环糊精(G2-β-CD)[7-12]、半乳糖基环糊精(Gal-β-CD)[13,14]、甘露糖基环糊精(Man-β-CD)[15]等已经通过酶法制备得到。
本课题组利用普鲁兰酶的反向合成特性[16],经过优化实验设计将麦芽三糖基引入到母体环糊精上获得了麦芽三糖基-β-环糊精(G3-β-CD),并对 G3-β-CD进行了结构分析和鉴定。
1 材料与方法
1.1 实验材料
G3-β-CD 是以普鲁兰和 β-环糊精为底物,应用普鲁兰酶水解和反向合成特性制备,并在本实验室进行分离纯化;普鲁兰、β-环糊精,上海 Sigma-Aldrich公司;普鲁兰酶,杰能科生物工程有限公司;其他试剂为国产分析纯。
1.2 实验仪器
HPLC-20AT,日本岛津公司;NEXUS47傅里叶变换红外光谱仪,美国Thermo公司;Waters ZMD电喷雾质谱仪,美国Waters公司;AVANCE 400核磁共振仪,德国Bruker公司;AB135-S型电子分析天平,梅特勒-托利多仪器有限公司;Delta320 pH计,梅特勒-托利多仪器有限公司。
1.3 实验方法
1.3.1 麦芽三糖基-β-环糊精的纯度分析
采用氨基柱进行分析,检测器为示差折光检测器,泵为岛津 LC-20A;色谱柱为 Lichrospher NH2(5 μm,4.6 mm ×250 mm);乙腈∶水(体积比 70∶30);柱温选择35°C;流速1 mL/min;进样量10 μL。
1.3.2 红外光谱分析方法
采用KBr压片法制备分析样品,干燥样品与KBr以1∶100的质量比进行混合研磨,压片后置于NEXUS47傅里叶变换红外光谱仪中,进行测试。
1.3.3 电喷雾质谱分析
采用电喷雾电离质谱分析样品分子质量。在Waters ZMD电喷雾电离质谱仪上,采用电喷雾式电离,电喷雾电压为3.7 kV,离子源的温度为120℃,脱溶温度为300℃,离子能量选用1.0eV。
1.3.4 麦芽三糖基-β-环糊精的酶解分析
将10 mg G3-β-CD 溶解于 0.5 mL 乙酸-乙酸钠缓冲溶液(20mmol/L、pH 5.0)中,加入普鲁兰酶,在50℃下孵育30 min,高温灭活酶后,对酶解产物进行HPLC分析。
1.3.5 核磁共振分析方法
将G3-β-CD溶解于重水后,放置于5 mm内径的专用核磁管中,在AVANCE 400核磁共振仪上进行的1H NMR,13C NMR和HMBC分析。
2 结果与讨论
2.1 麦芽三糖基-β-环糊精的纯度分析
如图1所示,由本实验室所制备分离纯化得到的G3-β-CD纯度很高,根据HPLC峰面积积分,其纯度>99.5%。这表明本课题组所采用的G3-β-CD制备分离方法科学有效,这为进一步确定其结构奠定了基础。

图1 G3-β-CD 的 HPLC 图Fig.1 HPLC of G3-β-CD
2.2 麦芽三糖基-β-环糊精的红外光谱分析
如图2所示,红外光谱曲线从上到下依次是G3-β-CD、β-CD和麦芽三糖(G3)。图2中3种成分的红外光谱都显示了糖类的特征吸收峰,3 400 cm-1为O—H的伸缩振动峰,2 927 cm-1为C—H伸缩振动峰,1 640 cm-1为 O—H 弯曲振动峰,1 150 cm-1为 CO振动峰,这与糖类结构解析文献中的报道一致[17]。图2中845~855 cm-1波长范围出现的吸收峰主要是由α-糖苷键的特征吸收所引起的,在红外谱图中未出现β-糖苷键的特征吸收峰,这表明麦芽三糖基是以α型糖苷键与β-CD相连。从图2中还可以看到,β-CD在3 400 cm-1左右的羟基吸收峰的峰形比G3-β-CD的峰形更宽,这主要是由于麦芽三糖基接枝到β-CD,部分地打开了环糊精分子内的氢键,致使峰形尖锐,而母体环糊精由于形成的分子内氢键稳定,化学键力常数小,从而在3 400 cm-1处产生宽而强的吸收。由于分子内羟基所形成的氢键是阻碍环糊精分子水合的主要原因,侧链基团麦芽三糖基的引入部分地破坏了母体环糊精的分子内氢键从而减少了分子水合阻碍,这有利于增加G3-β-CD在水中的溶解度。

图2 G3-β-CD 的红外光谱Fig.2 FTIR spectrum of G3-β-CD
2.3 麦芽三糖基-β-环糊精的电喷雾质谱
根据电喷雾电离质谱的一般规律,结合图3中[M-H]+和[M]-的 m/z分别为1 619.5和1 621.5,可推断出麦 G3-β-CD 的分子质量为 1 620.5,这与G3-β-CD的理论分子质量相一致。

图3 G3-β-CD 的电喷雾质谱Fig.3 ESIMS spectra of G3-β-CD in the negative and positive mode
2.4 麦芽三糖基-β-环糊精的酶解分析
为了进一步确证产品G3-β-CD的结构,采用普鲁兰酶水解G3-β-CD,酶解1 h后的产物经HPLC检测,并与麦芽三糖、β-CD标准品的HPLC图比较,发现酶解产物的HPLC保留时间分别为12.359 min、16.212 min,这与麦芽三糖和β-CD标准品的保留时间相一致。这说明本课题组所合成的G3-β-CD是由麦芽三糖和β-CD组成,结合电喷雾质谱分析的分子量,可以推断出G3-β-CD是由一分子麦芽三糖通过糖苷键与一分子β-CD相连接。
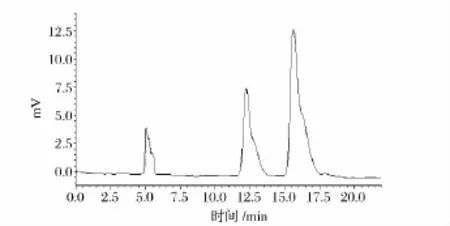
图4 G3-β-CD 酶解产物的 HPLCFig.4 HPLC of G3-β-CD hydrolyzed with pullulanase
2.5 麦芽三糖基-β-环糊精的核磁共振分析
G3-β-CD 的1H NMR 分析如图5所示,参照文献[18-20],对 G3-β-CD 的质子化学位移进行归属。1H NMR谱中包含3种不同的端基质子,分别为环糊精葡萄糖残基A1~A7上的H-1(δ 4.95-4.9)、接枝点葡萄糖残基B1上的的H-1(δ 4.8)和支链葡萄糖残基 B2和 B3对应的 H-1(δ 5.17,δ 5.23),3 种类型的端基质子强度积分比为7∶1∶2,这说明3种不同的葡萄糖残基摩尔比为7∶1∶2。
G3-β-CD的13C NMR 分析如图6所示,参照文献[18-21],对 G3-β-CD 的碳信号进行归属。G3-β-CD的13C NMR谱中包含3种不同的C-1信号,分别为环糊精葡萄糖残基 A1~A7的 C-1(δ 103.05-102.76),接枝点葡萄糖残基B1的C-1(δ 99.68)和支链葡萄糖B2和B3的C-1(δ 101.06-100.87),3种C-1的信号强度比为7∶1∶2。从13C NMR谱图发现了2种C-6信号,一种来自环糊精葡萄糖残基A1~A7和支链葡萄糖残基B2和B3的C-6信号(δ 61.68-61.25),一种来自接枝点葡萄糖残基B1的C-6信号(δ 68.20)。由于吡喃糖残基的C-6信号通常出现高磁场区域,δ 68.20出现的C-6信号明显地向低场发生了移动,这表明麦芽三糖基接枝在葡萄糖残基B1上。

图5 G3-β-CD 的1H 核磁共振图Fig.5 1H NMR spectrum of G3-β-CD

图6 G3-β-CD 的13C 核磁共振图Fig.6 13C NMR spectrum of G3-β-CD
G3-β-CD的13C NMR 谱有4种 C-4的信号,分别为环糊精分子 A1~A7上的 C-4(δ 82.64-82.04),接枝点葡萄糖残基B1的C-4(δ 79.18)、支链葡萄糖残基B2的C-4(δ 78.01)和支链最远端葡萄糖残基B3的C-4(δ 70.45),葡萄糖残基B3的C-4由于远离环糊精主环,化学位移受糖苷键影响较小,很大程度保持了其原来的化学位移值。
HMBC谱可以揭示糖单元的连接方式,G3-β-CD的HMBC谱如图7(a)所示。从图7(a)中可以看出,环糊精分子葡萄糖残基A1~A7的C-4与H-1在δ 82.32,5.06出现了多重耦合峰,C-1与H-4在δ103.02,3.58出现了多重耦合峰,表明产品中C-4与H-1、C-1与H-4相连,这表明环糊精分子的主环糖残基以α-1,4糖苷键首尾相连。HMBC谱中接枝点葡萄糖残基B1的C-6与环糊精葡萄糖残基A7的H-1在 δ 68.20,4.93出现单一耦合峰(图7(b)),这表明该葡萄糖残基 B1和A7以 α-1,6糖苷键相连。接枝点葡萄糖残基B1的C-4与支链葡萄糖残基B2的H-1在δ 79.18,5.32出现单一耦合峰(图7(c)),这表明葡萄糖残基 B1和 B2是以 α-1,4糖苷键相连。综合以上分析可以得出结论G3-β-CD的结构为单取代-6-O-α-D-麦芽三糖基-β-环糊精(图8)。
3 结论
经过 HPLC鉴定,G3-β-CD 的纯度达到 99%以上,这表明本课题组所设计的制备和分离方案科学有效。通过电喷雾质谱的分析,可计算出 G3-β-CD的分子质量为1 620.5,这与其理论分子质量相一致。结合酶解产物的HPLC分析和红外光谱,可知产物有一分子的麦芽三糖通过α型糖苷键与β-CD连接而成,红外谱图中麦芽三糖基-β-环糊精在3 400 cm-1左右的羟基吸收峰尖锐,这是由于糖基的引入后破坏了母体环糊精分子内氢键,增加了改性环糊精的溶解度。通过1H NMR,13C NMR和HMBC谱进一步确证了其结构为单取代-6-O-α-D-葡萄吡喃糖基-β-环糊精。

图7 G3-β-CD 的 HMBC核磁共振图Fig.7 HMBC spectrum of G3-β-CD

图8 G3-β-CD的HMBC核磁共振图Fig.8 HMBC spectrum of G3-β-CD
[1]金征宇,徐学明,陈寒青,等.环糊精化学—制备与应用[M].北京:化学工业出版社,2009:17-24.
[2]Tavornvipas S,Hirayama F,Arima H,et al.6-O-α-(4-O-α-D-glucuronyl)-D-glucosyl-β-cyclodextrin:solubilizing ability and some cellular effects[J].International Journal of Pharmaceutics,2002,249(1-2):199-209.
[3]Bellringer M E,Smith T G,Read R,et al.β-Cyclodextrin:52-week toxicity studies in the rat and dog[J].Food and Chemical Toxicology,1995,33(5):367-376.
[4]Abe J,Takeda Y,Hizukuri S,et al.Isolation and characterisation of 6-α-D-glucosylcyclomaltoheptaose[J].Carbohydrate Research,1984,131(1):175-179.
[5]Kobayashi S,Shibuya N,Yong M,et al.The preparation of 6-O-α-D-glucopyranosyl cyclohexaamylose [J].Carbohydrate Research,1984,126(2):215-224.
[6]Watanabe N,Yamamoto K,Tsuzuki W,et al.A novel method to produce branched-cyclodextrins:pullulanaseglucoamylase-mixed method [J].Journal of Fermentation and Bioengineering,1997,83(1):43-47.
[7]Shiraishi T,Kusano S,Sumuraya Y,et al.Synthesis of maltosyl(α,1-6)cyclodextrins the reverse reaction of thermostable Bacillus acidopullyticus pullulanase [J].Agricultural and Biological Chemistry,1989,153(1):2 181-2 188.
[8]王少杰,金征宇.普鲁兰酶逆向合成Mal-β-CD的工艺研究[J].食品工业科技,2005,26(9):105-107.
[9]张永伟,徐学明,赵建伟,等.普鲁兰酶逆向合成麦芽糖基-α-环糊精[J].食品与发酵工业,2009,35(6):46-49.
[10]崔波,金征宇.普鲁蓝酶逆向合成麦芽糖基β-环状糊精[J].食品科学,2005,26(12):128-131.
[11]崔波,金征宇.麦芽糖基(α-1→6)β-环糊精的酶法合成和结构鉴定[J].高等学校化学学报,2007,28(2):283-285.
[12]Yim D K,Park Y H.Production of branched cyclodextrins by reverse reaction of microbial debranching enzyme[J].Starch/Stärke,1997,49(2):75-78.
[13]Kitahata S,Hara K,Fujita K,et al.Synthesis of 6-O-α-D-galactosyl-α-cyclodextrin by coffee bean α – galactosidase [J].Bioscience Biotechnology and Biochemistry,1992,56(1):1 518-1 519.
[14]沈汪洋,金征宇,高虹.α-半乳糖苷酶法合成 α-半乳糖基-β-环糊精的研究[J]:湖北农业科学,2011,50(19):4 041-4 043.
[15]Ishiguro T,Fuse T,Oka M,et al.Synthesis of branched cyclomaltooligosaccharide carboxylic acids(cyclodextrin carboxylic acids)by microbial ocidation [J].Carbohydrate Research,2001,331(4):423-430.
[16]于博,吴月纯,金征宇,等.普鲁兰酶的反向合成特性[J].江苏农业科学,2012,40(12):31-34.
[17]Tao Y,Zhang L.Characterization of polysaccharide-protein complexes by size-exclusion chromato-grammphy combined with three detectors [J].Carbohydrate Research,2008,343(13):2 251-2 257.
[18]Ishizuka Y,Nemoto T,Kanazawa K,et al.1H NMR spectra of branched-chain cyclomaltohexaoses(α-cyclodextrins)[J].Carbohydrate Research,2004,339(4):777-785.
[19]Okada Y,Semma M,Ichikawa A.Physicochemical and biological properties of 61,63,65-tri-O-α-maltosyl-cyclomaltoheptaose(61,63,65-tri-O-α-maltosyl-β-cycylodextrin)[J].Carbohydrate Research,2007,342(10):1 315-1 322.
[20]Cui B,Jin Z.Enzymatic synthesis and identification of maltosyl(α-1→6)β-cycylodextrin [J].Chemical Journal of Chinese University,2007,28(2):283-285.
[21]Bock K.Carbohydrate-protein interactions:substrate specificity of enzymes used in the degradation of oligosaccharides related to starch and cellulose.Pure and Applied Chemistry,1987,59(11):1 447-1 456.
