Nrf2基因敲除对肝脏氧化应激及胰岛素抵抗的影响
2013-11-15闻勤生赵曙光王旭霞王景杰刘震雄第四军医大学唐都医院消化内科西安710038通讯作者mailliuzx816yahoocomcn
倪 阵,闻勤生,赵曙光,张 哲,王旭霞,王景杰,刘震雄(第四军医大学唐都医院消化内科,西安 710038;通讯作者,E-mail:liuzx816@yahoo.com.cn)
非酒精性脂肪性肝炎(NASH)是全球范围内最常见的慢性肝脏疾病之一,被认为是代谢综合征在肝脏的表现[1]。胰岛素抵抗(IR)和NASH关系密切,并能通过促进肝脏脂肪变性、细胞损伤和炎症反应,最终导致非酒精性单纯性脂肪肝(NAFL)向非酒精性脂肪性肝炎(NASH)的发展[2],而氧化应激是诱导IR的重要危险因素,其可以直接或间接活化多种应激通路,进一步增加胰岛素受体底物-1(IRS-1)不连续的丝氨酸或苏氨酸磷酸化,从而抑制IRS-1酪氨酸磷酸化,最终导致IR的发生[3]。核转录因子NF-E2相关因子2(Nrf2)是体内调节氧化应激的关键靶点,其可以通过促进多种Ⅱ相抗氧化酶和解毒酶的表达[4],发挥重要的抗氧化作用,但Nrf2在肝脏IR中的作用尚未见明确报道。本实验将Nrf2基因敲除小鼠作为研究对象,通过给予4周高脂饮食,观察肝脏氧化应激及IR相关指标的变化,直接探讨Nrf2在肝脏IR发生中的作用。
1 试剂和方法
1.1 试剂
牛胆盐、胆固醇均购自西安昕泰生物技术责任有限公司;血糖、丙二醛(MDA)与谷胱甘肽(GSH)检测试剂盒,BCA蛋白定量试剂盒均购自南京建成生物有限公司;兔抗鼠JNK多克隆抗体、兔抗鼠p-JNK多克隆抗体、兔抗鼠IRS-1多克隆抗体、兔抗鼠p-IRS-1多克隆抗体、β-actin均购自英国Abcam公司;羊抗兔IgG抗体、β-actin二抗均购自北京博奥森生物有限公司。
1.2 动物来源及分组
清洁级ICR雄性野生型(WT)和Nrf2基因敲除(Nrf2-/-)小鼠各10只,6-8周龄,体重18-22 g,购自南京军区总医院比较医学科。所有小鼠适应性喂养1周后,按照随机数字法分为WT对照组(control)、Nrf2-/-对照组(KO)、WT 高脂饮食组(HFD)和Nrf2-/-高脂饮食组(KOHFD),各5只。对照组给予普通饲料,由第四军医大学实验动物中心提供,高脂饮食组给予高脂饲料(在普通饲料基础上添加10%猪油、2%胆固醇和0.5%胆盐),由西安迪乐普生物有限公司生产加工,每周记录小鼠体重变化。4周末,空腹12 h,行腹腔注射葡萄糖耐量实验,之后4%戊巴比妥钠腹腔注射麻醉,下腔静脉取血,肝脏称重,部分固定于4%多聚甲醛,行组织学检测,其余冻存于液氮中,留待检测。
1.3 检测指标及方法
1.3.1 空腹血糖检测 下腔静脉采血,室温静置2 h,2 000 r/min离心10 min,吸取上清,按照血糖检测试剂盒说明书检测空腹血糖变化。
1.3.2 葡萄糖耐量实验(iPGTT)所有小鼠空腹12 h后,称体重,腹腔注射20%葡萄糖溶液(2 g/kg),分别在 0 min,15 min,30 min,60 min 和 120 min,尾静脉采血,检测血糖变化并进一步计算时间-血糖曲线下面积(AUC),公式为:

1.3.3 肝脏MDA和GSH检测 精确称取肝脏组织0.5 g,加入 4.5 ml预冷 PBS,手动匀浆 8 min,2 500 r/min离心10 min,取上清,按照试剂盒说明书检测MDA和GSH水平。
1.3.4 Western-Blot检测肝脏 JNK、p-JNK、IRS-1、p-IRS-1水平 取肝组织,研磨,裂解,提取蛋白并定量,进行聚丙烯酰胺凝胶电泳90 min,按照湿转法将电泳产物转移至NC膜上,5%脱脂奶粉封闭,4℃过夜,滴加一抗(1∶400),室温 4 h,PBST 洗膜 3 次,10 min/次,然后滴加二抗(1∶2 000),室温下孵育1 h,PBST洗膜3次,10 min/次。滴加 NBT/BCIP显色液,避光显色10 min,双蒸水洗膜终止显色,GelDoc凝胶成像仪采集图像。
1.3.5 肝脏组织学检测 取肝右叶部分组织,4%多聚甲醛固定24 h,石蜡包埋,切片,行苏木精-伊红染色,光镜下观察肝脏组织学改变。
1.4 统计学分析
2 结果
2.1 生理指标变化
HFD组和KOHFD组小鼠体重增加均高于control组和KO组,但差异均无统计学意义(P>0.05);与control组和KO组相比,KOHFD组小鼠肝脏质量显著升高(P<0.05),而HFD组小鼠肝脏质量与control组之间差异无统计学意义(P>0.05,见表1)。
2.2 肝脏光镜下改变
HE染色显示,control组和KO组小鼠肝脏肝小叶结构正常,肝细胞排列成条索状,围绕在中央静脉周围,呈放射状分布;HFD组小鼠肝脏组织结构较对照组明显紊乱,肝细胞内可见少量脂肪沉积,无明显炎症细胞浸润;而KOHFD组小鼠肝脏,脂肪沉积明显加重,部分细胞出现空泡变性,并可见少量炎症细胞浸润,主要位于静脉周围(见图1)。
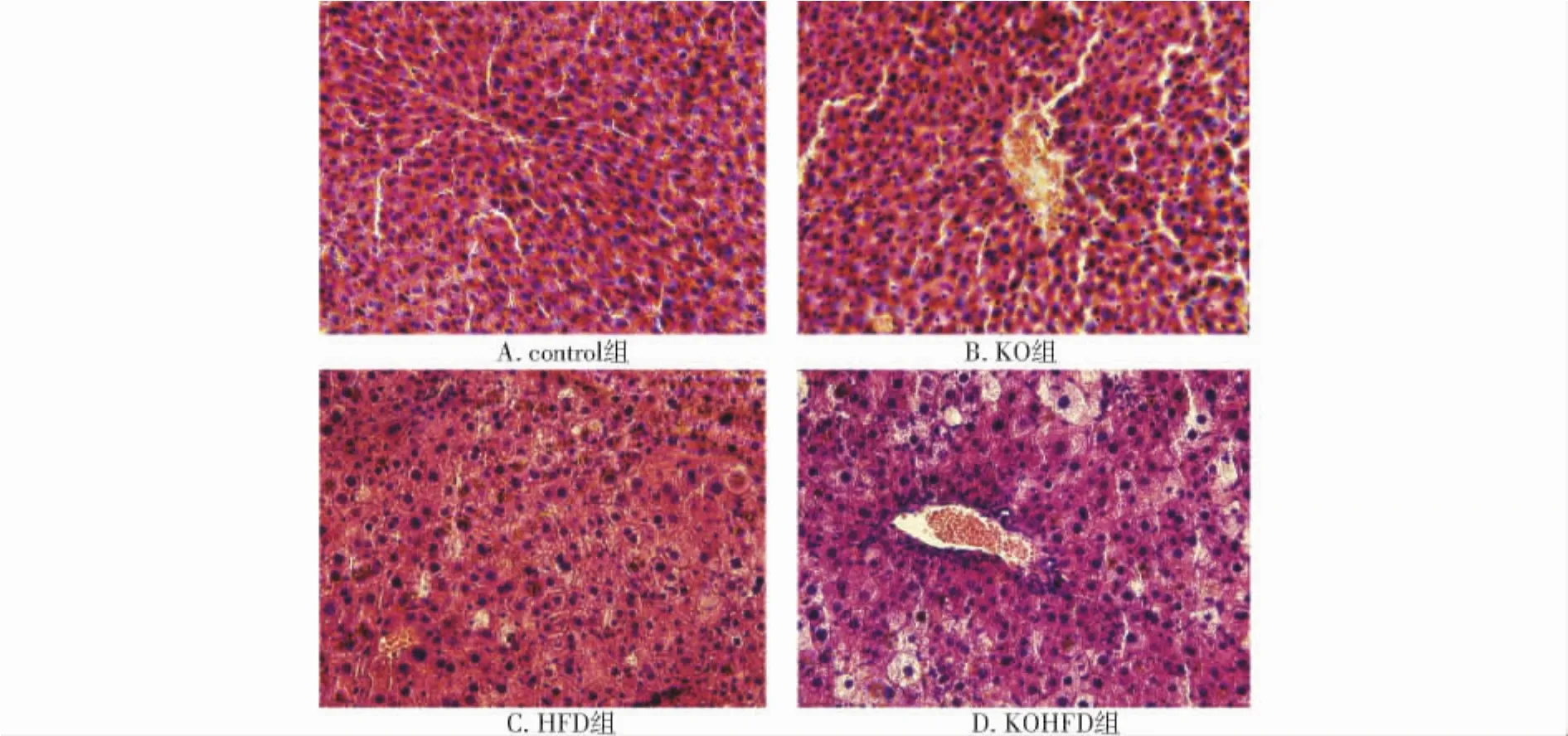
图1 各组小鼠肝脏组织学改变 (×40)Fig 1 Changes of pathology of liver in four groups (×40)
2.3 肝脏氧化应激水平变化
HFD组和KOHFD组小鼠肝脏丙二醛水平均显著高于control组和KO组(P<0.05),且KOHFD组明显高于HFD组(P<0.05);相反,与control组和KO组相比,HFD和KOHFD组小鼠肝脏谷胱甘肽水平均显著降低(P<0.05),且 KOHFD组明显低于HFD 组(P<0.05,见表1)。

表1各组小鼠体重、肝脏重量、空腹血糖、肝脏GSH和MDA变化(x¯±s)Tab 1The changes of weight gain,liver weight,blood glucose,hepatic GSH and MDA in mice(x¯±s)
2.4 空腹血糖变化
KOHFD组和HFD组小鼠空腹血糖均高于control组和KO组,但差异均无统计学意义(P>0.05,见表1)。
2.5 葡萄糖耐量实验变化
腹腔注射葡萄糖后,HFD和KOHFD组小鼠血糖水平在15 min,30 min和60 min均高于control组和KO组,且KOHFD组小鼠15 min血糖水平显著升高(P<0.05),而HFD组和control组之间差异无统计学意义(P>0.05);KOHFD组小鼠时间-血糖曲线下面积(AUC)显著高于control组(P<0.05),而HFD组与control组之间差异无统计学意义(P>0.05,见图2)。
2.6 肝脏JNK/p-JNK、IRS-1/p-IRS-1表达变化
各组小鼠肝脏JNK和IRS-1蛋白表达水平均无明显差异;p-JNK表达水平在KOHFD组小鼠肝脏明显升高,灰度值显著高于 KO组(P<0.05),而在HFD组小鼠肝脏表达轻度升高,灰度值与control组相比无显著差异(P>0.05);KOHFD组小鼠肝脏p-IRS-1表达显著减少,灰度值明显低于KO组(P<0.05),而HFD组小鼠肝脏p-IRS-1表达轻度下降,灰度值与control组之间无明显差异(P>0.05,见表2、图 3)。
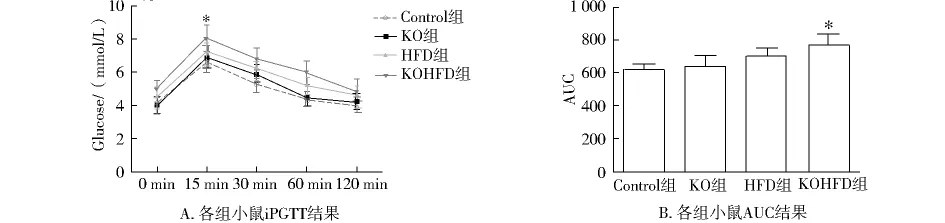
图2 各组小鼠葡萄糖耐量实验(iPGTT)和时间-血糖曲线下面积(AUC)Fig 2 The iPGTT and AUC results in four groups
表2肝脏JNK/p-JNK、IRS-1/p-IRS-1表达灰度值变化(±s)Tab 2The changes of grey value density of liver JNK/p-JNK,IRS-1/p-IRS-1 expression in mice(±s)

表2肝脏JNK/p-JNK、IRS-1/p-IRS-1表达灰度值变化(±s)Tab 2The changes of grey value density of liver JNK/p-JNK,IRS-1/p-IRS-1 expression in mice(±s)
与 control组比较,﹡ P <0.05;与 KO 组比较,△P <0.05
组别JNK p-JNK IRS-1 p-IRS-1 control组 1.573 ±0.132 1.436 ±0.096 1.689 ±0.087 1.613 ±0.167 KO 组 1.493 ±0.142 1.412 ±0.098 1.703 ±0.127 1.598 ±0.150 HFD 组 1.524 ±0.145 1.699 ±0.128 1.673 ±0.148 1.382 ±0.089 KOHFD 组 1.508 ±0.161 2.076 ±0.162﹡△ 1.681 ±0.189 1.003 ±0.079﹡△
3 讨论

图3 Western blot检测肝脏 JNK/p-JNK、IRS-1/p-IRS-1表达变化Fig 3 Expression of JNK/p-JNK,IRS-1/p-IRS-1 in livers by Western blot
非酒精性脂肪性肝病(NAFLD)是一种在病理学上与酒精性肝病相似,但患者无过量饮酒史的代谢性疾病,其疾病谱包括单纯性脂肪肝和脂肪性肝炎(NASH)。流行病学调查显示,其在全世界成人范围内的发病率约为17%-30%[5,6]。目前观点认为,单纯性脂肪肝属于良性病变,而NASH则可以进展至肝硬化、终末期肝病和肝癌。
本实验肝脏组织学检测显示,control与KO小鼠肝脏小叶结构清晰,肝细胞排列成条索状。给予4周高脂饮食后,HFD小鼠表现为轻度的肝脏脂肪变性,无明显炎症细胞浸润,而KOHFD小鼠肝脏脂质沉积较HFD小鼠明显加重,并显示出少量的炎症细胞浸润。这些结果说明,单纯敲除Nrf2对小鼠肝脏组织学无明显影响,而敲除Nrf2可以明显加重高脂饮食诱导下小鼠肝脏脂肪沉积和炎症变化。
氧化应激是促使单纯性脂肪肝向NASH进展的关键因素,当过量的活性氧(ROS)作用于生物膜上的多聚不饱和脂肪酸(PUFAs)时,引发脂质过氧化反应,从而使细胞内生物膜的通透性和流动性发生改变,导致细胞结构改变和功能障碍[7]。但是,机体只有在抗氧化能力下降的状态下,氧化应激才能产生负面作用。丙二醛(MDA)是细胞毒性较强的脂质过氧化产物,它半衰期较长,并能够弥散到细胞内其他靶位,加重氧化应激损伤。核转录因子NFE2相关因子2(Nrf2)是体内调节氧化应激的中枢环节,正常情况下,其与Keap1结合,主要位于细胞质内,处于相对抑制状态,当受到外源性或内源性刺激后,Nrf2从Keap1中释放,转位进入细胞核,与抗氧化反应元件(ARE)结合,从而调节靶基因的转录[8],表达具有抗氧化作用的Ⅱ相解毒酶。谷胱甘肽(GSH)是一种重要的内源性抗氧化产物,可以对抗自由基对细胞和器官的伤害[9]。Nrf2可以通过调节谷氨酸半胱氨酸合成酶(GCLC)和谷氨酸半胱氨酸合成酶调节亚单位(GCLM),调控GSH的合成[10]。我们前期的研究证实[11-13],通过诱导 Nrf2核转位,可以明显降低肝细胞和高脂饮食诱导的氧化应激水平。本实验结果显示,与control小鼠相比,KO小鼠肝脏MDA及GSH水平均无明显变化,而4周高脂饮食后,HFD小鼠肝脏MDA水平显著升高,而GSH水平显著下降,说明HFD小鼠肝脏已经产生氧化应激,而KOHFD小鼠MDA明显高于HFD小鼠,GSH水平明显低于HFD小鼠。说明单纯敲除Nrf2对小鼠肝脏氧化应激水平无明显影响,4周高脂饮食可以导致小鼠肝脏氧化应激的发生,而敲除Nrf2后,小鼠肝脏氧化应激水平进一步加重。
NASH患者往往存在胰岛素抵抗,大量实验也发现,高脂饮食诱导的NASH动物模型,伴有不同程度的高血糖、高胰岛素血症和胰岛素敏感性下降[14]。肝脏是胰岛素作用的靶器官,当存在高胰岛素血症时,肝脏葡萄糖输出持续存在而加重高血糖,反之,高血糖则可以进一步刺激胰岛素分泌,从而加重胰岛素抵抗,形成恶性循环。胰岛素抵抗可以促进外周脂肪的水解,导致游离脂肪酸(FFA)向肝脏运输增加,同时,高胰岛素血症可以刺激肝脏的脂肪合成,从而导致脂质在肝脏大量沉积[15]。
最近有研究提示,高脂饮食诱导的NASH小鼠,肝脏是首要受损器官,肝脏胰岛素抵抗先于外周胰岛素抵抗的发生[16],因此,肝脏IR是NASH进展的启动环节,而肝脏局部氧化应激的产生可能是诱发肝脏胰岛素抵抗的关键环节。其可以激活一系列信号通路,如JNK,而磷酸化的JNK则可以作用于胰岛素受体底物(IRS-1),促进其丝氨酸磷酸化、抑制其酪氨酸磷酸化,从而抑制胰岛素信号的转导[17]。本实验结果显示,control组和KO组小鼠空腹血糖、iPGTT和AUC均无明显差异,4周高脂饮食后,虽然HFD小鼠空腹血糖、时间-血糖曲线下面积(AUC)均高于control组,但差异无统计学意义。Western blot检测发现,control组和KO组小鼠p-JNK与p-IRS-1无明显差异,HFD小鼠肝脏p-JNK与p-IRS-1水平和control组相比轻度升高,但灰度值差异无统计学意义,这些结果说明,HFD小鼠没有出现明显的肝脏和外周胰岛素抵抗。而在KOHFD小鼠,虽然空腹血糖与control与KO小鼠相比无统计学意义,但是葡萄糖耐量实验结果显示,15 min血糖水平和AUC显著高于control组,说明KOHFD小鼠虽没有发生高血糖,但是已经存在轻度外周胰岛素抵抗。进一步Western blot检测提示,JNK和IRS-1水平在各组间无明显差异,而KOHFD小鼠肝脏p-JNK显著升高,p-IRS-1水平显著降低,说明KOHFD小鼠已经出现了显著的肝脏胰岛素抵抗。这些结果证实,单纯敲除Nrf2对小鼠葡萄糖代谢及肝脏胰岛素信号通路无明显影响,而给予敲除Nrf2小鼠高脂饮食可以通过加重肝脏氧化应激水平,进一步作用于胰岛素信号通路,明显加速肝脏胰岛素抵抗的发生。
本实验采用Nrf2基因敲除小鼠作为研究对象,通过给予高脂饮食,发现敲除Nrf2可以促进肝脏脂质沉积和炎症细胞浸润,加重肝脏氧化应激水平,进而增加JNK磷酸化水平、降低IRS-1酪氨酸磷酸化水平而显著加速小鼠肝脏胰岛素抵抗的发生。本实验的研究结果为以Nrf2为靶点治疗NASH相关胰岛素抵抗提供了新的实验依据。
[1] Farrell GC,Larter CZ.Nonalcoholic fatty liver disease:from steatosis to cirrhosis[J].Hepatology,2006,43(2 Suppl 1):p.S99-S112.
[2] Lyn PA.Nonalcoholic fatty liver disease:relationship to insulin sensitivity and oxidative stress[J].Altern Med Rev,2002,7(4):276-291.
[3] Beyer TA,Xu W,Teupser D,et al.Impaired liver regeneration in Nrf2 kockout mice-role of ROS-mediated insulin/IGF-1 resistance[J].EMBO J,2008,27(1):212-223.
[4] Zhang Q,Pi JB,Courtney G.Woodset al.A systems biology perspective on Nrf2-mediated antioxidant response[J].Toxicol Appl Pharmacol,2010,244:84-97.
[5] Raj VU,Naga CH.Non-alcoholic fatty liver disease and non-alcoholic steatohepatitis:Selected practical issues in their evaluation and management[J].Hepatology,2009,49(1):306-317.
[6] Cheung O,Sanyal AJ.Recent advances in nonalcoholic fatty liver disease[J].Curr Opin Gastroenterol,2010,26:202-208.
[7] Giovanni MU,Roberto GA,Maurizio CA.Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver[J].Prog Lipid Res,2009,48:1-26.
[8] Roberto GA,Giovanni MU,Maurizio CA.Redox balance in the pathogenesis of nonalcoholic fatty liver disease:mechanisms and therapeutic opportunities[J].Antioxid Redox Signal,2011,15(5):1325-1352.
[9] Li WG,Kong AN.Molecular mechanisms of Nrf2-mediated antioxidant response[J].Mol Carcinog,2009,48(2):91-104.
[10] Hirokazu SU,Kosuke OK,Junichi SH,et al.Deletion of nuclear factor-e2-related factor-2 leads to rapid onset and progression of nutritional dteatohepatitis in mice[J].Am J Physiol Gastrointest Liver Physiol,2009,19.
[11] 赵丽,赵曙光,李慧艳,等.Nrf2对高脂饮食诱导的大鼠胰岛素抵抗的保护作用[J].山西医科大学学报,2011,42(9):723-726.
[12] 张晓兰,王旭霞,冯国华,等.网络抗氧化剂对大鼠非酒精性脂肪肝炎模型的防治作用[J].肝脏,2008,1(3):130-132.
[13] 李强,赵曙光,闻勤生,等.姜黄素激活转录因子Nrf2对人肝细胞氧化应激的影响[J].胃肠病学和肝病学杂志,2011,l9(2):154-156.
[14] Chavez-Tapia NC,Rosso1 N,Tiribelli C.In vitro models for the study of nonalcoholic fatty liver disease[J].Curr Med Chem,2011,18,1079-1084.
[15] Postic CA,Girard JE.Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance:lessons from genetically engineered mice[J].J Clin Invest,2008(118):829-838.
[16] Patrice DC,Jacques AM,Miguel AIet al.Metabolic endotoxemia initiates obesity and insulin resistance[J].Diabetes,2007,56:1761-1772.
[17] Samuel VT,Liu ZX,Qu XQ,et al.Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease[J].J Biol Chem,2004,279(31):32345-32353.
