宏基因组学在人和动物胃肠道微生物研究中的应用进展
2013-11-12许波杨云娟李俊俊唐湘华慕跃林黄遵锡
许波,杨云娟,李俊俊,唐湘华,慕跃林,黄遵锡
1 生物能源持续开发利用教育部工程研究中心,云南 昆明 650500
2 云南师范大学生命科学学院,云南 昆明 650500
3 云南省生物质能与环境生物技术重点实验室,云南 昆明 650500
4 云南师范大学酶工程重点实验室,云南 昆明 650500
人和动物胃肠道内存在大量微生物,它们与宿主的生理功能密切相关,这些微生物群落之间及微生物与动物宿主之间形成了相互依存、相互作用的不可分割的整体。正常状态下菌群和宿主之间相互交换能量物质、传递信息,对宿主有营养、免疫、刺激生长和生物颉颃等作用,在胃肠道系统中起重要作用[1]。胃肠道中的内源性微生物类群在种类、数量和定位等方面保持相对稳定,形成了胃肠道的微生态平衡,是人和动物机体内环境中不可缺少的组成部分。最近由我国科学家参与完成的人体肠道宏基因组研究计划 (European Metagenomics of the Human Intestinal Tract,MetaHIT)证明,人体肠道细菌群约有 300万个基因,是人体基因数量的约150倍[2]。这对诠释肠道细菌和人体健康的关系,研究肠道细菌在疾病发生中的作用,监控、预防、干预肠道细菌,开发新药都具有重要意义。因此,一个研究人体及动物胃肠道微生物的高潮正在兴起,本文旨在阐述宏基因组学在人和动物胃肠道微生物研究中的应用,同时着重介绍宏基因组研究的生物信息学技术。
1 宏基因组学简介
“Metagenome”最早是1998年由Handelsman等[3]提出的,其定义为“the collective genomes of soil microflora”,即生境中全部微小生物遗传物质的总和,既包含了可培养的又包含了未可培养的微生物基因,可翻译为宏基因组、元基因组或环境基因组等。在宏基因组学研究中,借助于大规模测序,结合生物信息学工具,能够发现大量过去无法获得的未知微生物新基因或新的基因簇,这对了解复杂环境中的微生物群落组成及其代谢功能,挖掘具有应用潜力的新基因具有重要意义。宏基因组学的研究通常有以下3种策略 (图1):1)利用引物PCR扩增16S rDNA (特别是V6区)或其他遗传标记,然后对扩增产物测序;2)构建宏基因组文库;3)直接进行Shotgun测序。
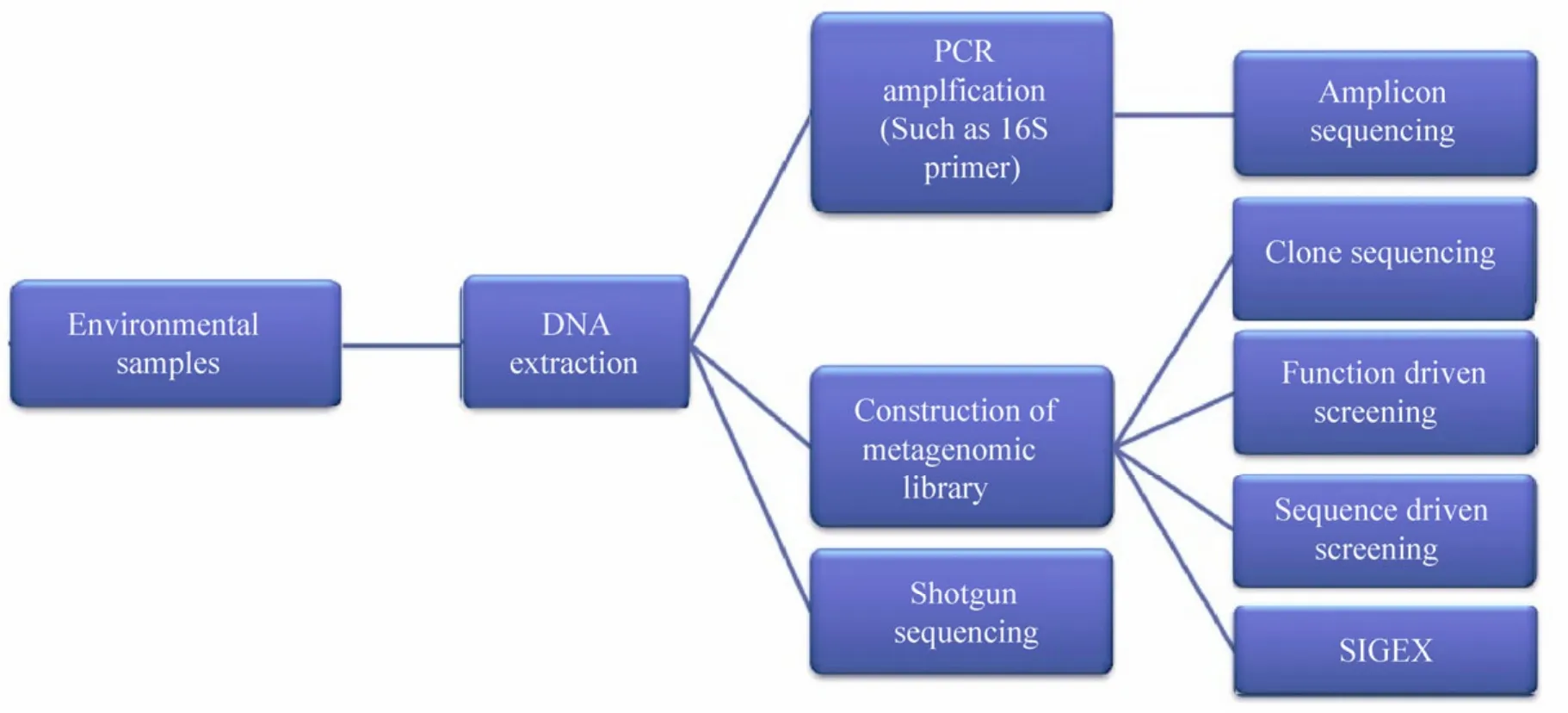
图1 宏基因组学的研究策略示意图Fig. 1 Possible steps in a metagenomic workflow.
2 宏基因组研究的生物信息学技术和平台
宏基因组学技术的兴起有效地解决了传统微生物学在检测和鉴定微生物物种方面的局限性,然而宏基因组学在发展过程中也遇到了与基因组学研究类似的瓶颈, 即如何高效分析宏基因组测序产生的海量数据,因此相关的生物信息学分析方法和平台就成为宏基因组学研究的重要手段。近年来,很多与宏基因组研究有关 (如数据的组装、基因预测、功能注释等)的计算系统和软件平台不断被开发,并被用于环境样品微生物宏基因组的物种分类和功能组成研究。
2.1 宏基因组组装
由于新一代测序技术的读长较短 (几十至几百个碱基对),只能覆盖基因的一部分,为此要将这些读段组装成较长的序列, 以重建全长基因,便于后续的分析处理。宏基因组组装即把 reads拼装成 contigs, 再由 contigs拼装成scaffolds,常用的软件见表1。主要可以分为两类:1)基于 overlap graph的拼接软件,包括Celera Assembler、ARACHNE、PCAP 等。2)基于de bruijn graph的拼接软件,包括SOAPdenovo、Velvet、ALLPATHS、ABySS、MOCAT 等。
在有足够高覆盖率的宏基因组测序中,通过序列组装有时可以重建群落中微生物的完整基因组[4-5]。然而 DNA保守区域的存在、微生物的变异和基因的水平转移,实际上会导致许多高度复杂的群落中出现嵌合体以及 scaffolds重建时产生歧义[6-7]。尽管存在这些理论上的局限,Qin 等[2]以及 The Human Microbiome Project Consortium[8]利用单基因组装配方法 (如SOAPdenovo),在从宏基因组中重建高丰度微生物群落时已取得了不错的效果。然而,近年来通过从群落中存在歧义的序列中剔除嵌合序列,研究者开发出了一些专门针对宏基因组的装配软件,例如 MetaVelvet、khmer、Meta-AMOS和Meta-IDBA等。这些软件采用基于图形的重建方法,有效解决了多重基因组中保守序列所引起的基因组拷贝数变化及组装时歧义序列的产生,因此被认为是研究复杂生态群落微生物组的有力工具。
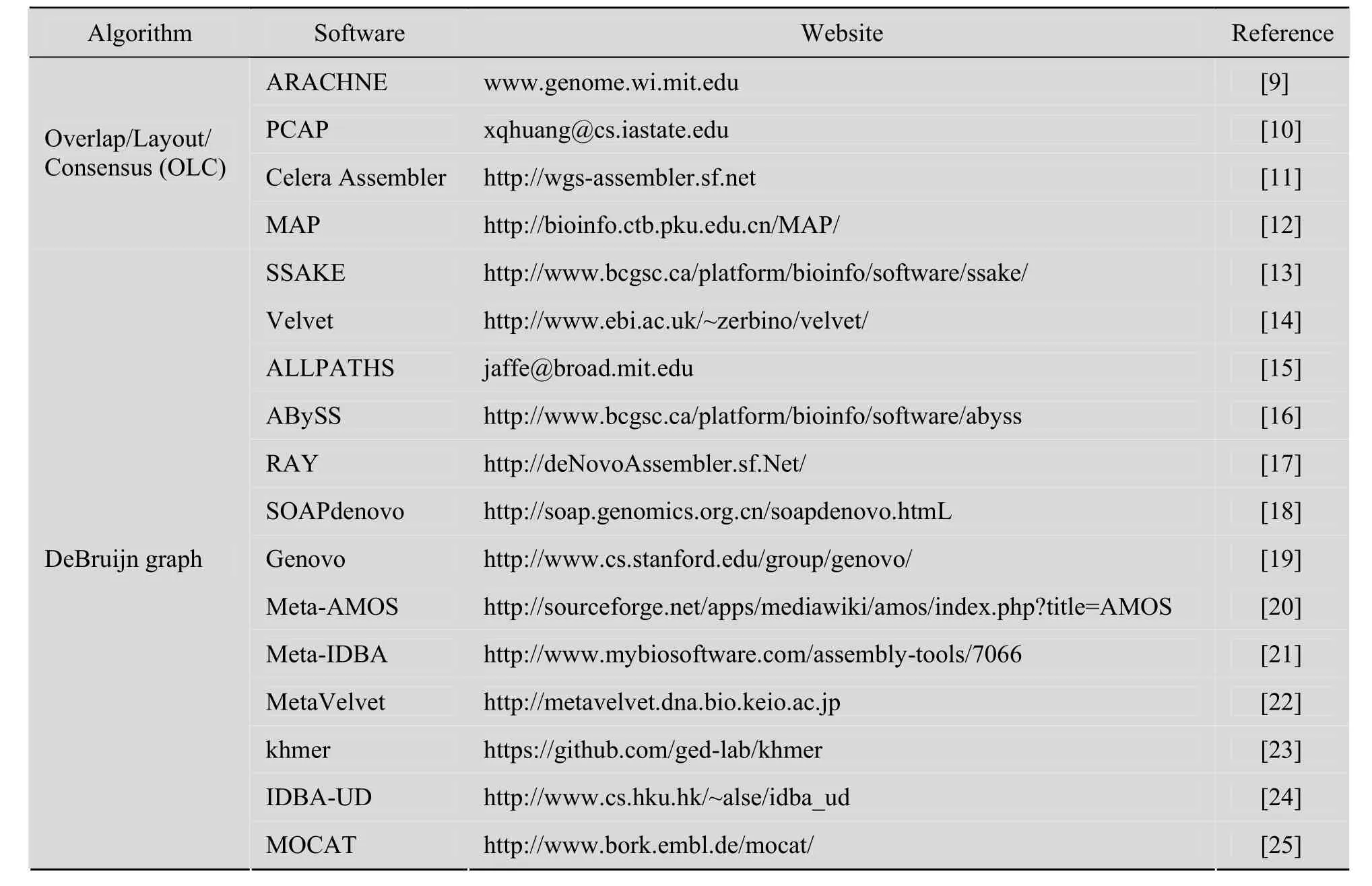
表1 宏基因组数据组装常用软件Table 1 Common software for metagenome assembly
2.2 基因预测
可用于宏基因组的基因预测软件如表 2所示,由于采用了适合短reads的统计学模型,因此能检测到不完整的ORFs,使得这些软件对基因的预测比传统基因预测方法更为灵敏和精确。然而宏基因组是由来自不同微生物的混合序列组成,不论用哪种方法对微生物宏基因组测序的数据进行基因预测,质量都不及对基因组测序数据的预测。为了弥补这些缺陷、优化预测结果,将几种基因预测方法合并起来使用是常用的策略[26]。
2.3 物种分类鉴定
将获得的序列及其基因功能分配至生境中特定微生物类群是当前宏基因组学研究的重要问题之一,然而群落内不同的物种丰度、复杂性,以及新物种的发现,使得对短读长的DNA序列进行物种分类极具挑战性。目前,研究者已开发出多种用于宏基因组物种分类鉴定的软件和工具 (表 3),总体说来可以分为两类[27]:1) 以序列内在特性为基础的分级方法(Intrinsic binning approaches):通常从参照基因组中选择系统发育分类器 (如 GC含量、k-mer频率等),然后利用统计学模型如Support Vector Machines with structured output (PhyloPythiaS)、interpolated Markov models (Phymm)、naive
Bayesian classifiers (NBC)、Self Organizing Maps(TaxSOM)等对宏基因组数据进行分级;2)以序列外部信息或同源性为基础的分类方法(Extrinsic or homology-based classification):该方法是将宏基因组序列与从参照基因组中提取的最为有效的特征标记 (如16S rRNA基因)直接进行比较,此外,将其他一些保守性高的基因纳入到这些通用标记物中,可进一步提高分类的普遍性和系统发育分辨率,例如AMPHORA[51]采用了31个标记物,其中主要是核糖体蛋白。此外,有的工具则是将这两种方法整合起来,如PhymmBL[43]、RITA[44]。
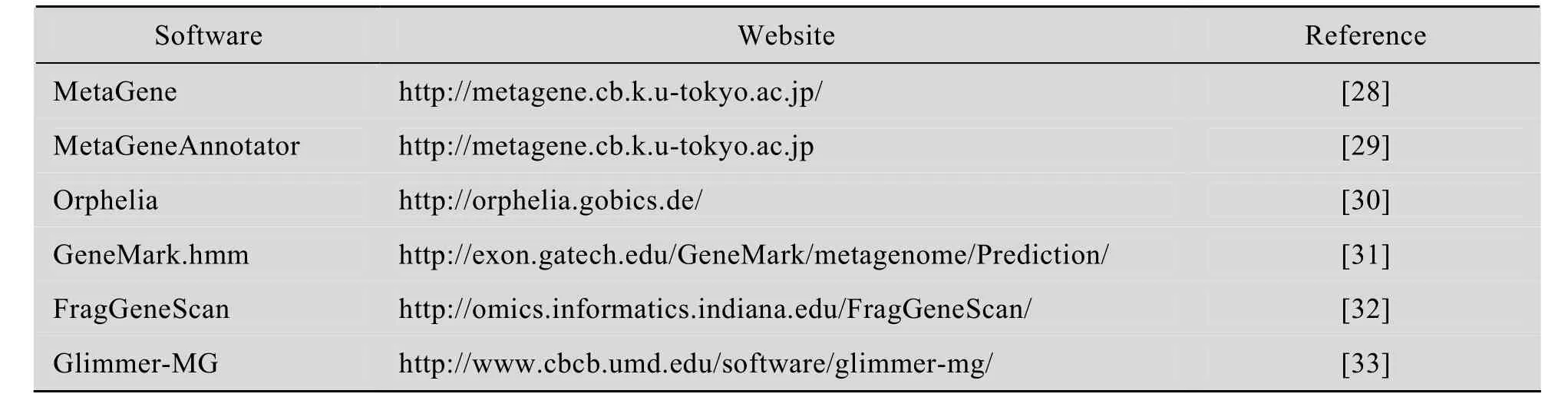
表2 宏基因组基因预测常用软件Table 2 Common software for gene prediction

表3 宏基因组物种分类鉴定常用工具Table 3 Common tools for taxonomic classification
2.4 功能注释
宏基因组的分析涉及通过与数据库比对来对预测的基因进行功能注释,例如对SwissProt、NCBI nr、KEGG或 COGs数据库进行蛋白质 Blast比对,对 Pfam和 TIGRfam数据库进行HMMer比对,或是针对一些具有特殊功能的数据库,如碳水化合物活性酶类数据库CAZy进行比对,注释的结果可以作为后续代谢重构等功能数据挖掘的基础。目前,一些专用的宏基因组注释平台和系统已经形成(表 4),例如 MG-RAST、IMG/M 和 WebMGA,这些平台不仅仅是单纯的注释系统,还陆续增加了一些如生物多样性分析、分类鉴定和宏基因组比较等功能。随后,一些专门针对宏基因组的比较工具也得到了开发,包括RAMMCAP、STAMP、METAREP、CoMet和METAGENassist。

表4 宏基因组基因功能注释常用工具Table 4 Common tools for metagenomic functional annotation
3 宏基因组学在动物胃肠道微生物研究中的应用
3.1 动物胃肠道微生物群落结构及多样性分析
地球上存在超过 55个细菌门类,但只有Bacteroidetes和Firmicutes这两个深层进化门类的菌群在胃肠道中占优势,这表明动物胃肠道是一个相对独立的环境,其微生物组成与其他生态环境有明显差别。
宏基因组研究表明,人类的胃肠道微生物组比人类的基因组显著富集与多糖、氨基酸和外源性化学物质代谢有关的基因[62],因此,胃肠道微生物组具有动物宿主不需要独立进化、而又能帮助宿主从食物中获取营养的基因,否则宿主就很难消化食物。同样,虽然大熊猫以富含纤维素的竹子为食,但其基因组中却缺乏消化纤维素的相关酶类基因,Zhu等通过分析大熊猫肠道微生物宏基因组,发现了包括纤维素酶、β-葡萄糖苷酶等在内的多种糖苷水解酶编码基因[63]。这些研究更进一步证实了胃肠道微生物与其宿主之间的共同进化,不同的宿主,由于饮食、遗传、环境和疾病等的影响都会在其进化过程中形成与自身协调发展的肠道微生物组,并具有编码相应酶类的基因,以适应宿主的生活状态。因此,在利用宏基因组技术对人类胃肠道微生物群落结构及多样性进行了大量系统的研究后[62-64],研究者开始关注与人类生活关系密切的动物,如鸡[65]、狗[66]、猪[67]和猫[68],以及牛[69-70]、鹿[71]和骆驼[72]等食草性动物的胃肠道微生物。
2011年,Swanson等对狗粪便微生物的宏基因组进行随机测序[66],分析发现Bacteroidetes/ Chlorobi group和Firmicutes是优势菌群,分别占所分析序列的约 35%,其次是Proteobacteria (13%~15%)和Fusobacteria (7%~8%),与其他动物胃肠道微生物宏基因组的层次聚类表明,狗与人类、鼠在系统分类和功能代谢上都最为相似。对猪粪便微生物宏基因组进行shotgun测序的研究表明[67],猪粪便微生物中占主导地位的是Firmicutes和Bacteroidetes,其中Prevotella spp.占优势,与其他动物宏基因组数据的比较发现,猪粪便菌群的门类分布与牛瘤胃和鸡盲肠最为相似。瘤胃中具有与纤维素降解密切相关的微生物群落,结合16S rRNA基因V3-V5区的扩增子测序及宏基因组随机测序研究表明[70],出生 42 d牛犊的瘤胃菌群中Bacteroidetes占优势 (74.8%),其次是Firmicutes(12.0%)、Proteobacteria (10.4%)、Verrucomicrobia(1.2%)和Synergistetes (1.1%)。而对骆驼瘤胃微生物宏基因组的比较分析也表明[72],由于骆驼与牛具有相似的消化道结构和功能,其瘤胃微生物群落的结构组成和功能都最为相似。
由于非人灵长类动物在进化上与人类关系较近,本实验室以一种低等灵长类动物倭蜂猴为研究对象,利用宏基因组随机测序对其粪便微生物的结构组成及其潜在功能进行了全面研究。物种组成分析表明,与人类相似,倭蜂猴粪便微生物组中Bacteroidetes、Proteobacteria、Actinobacteria和 Firmicutes占优势,然而其所占相对比例却不同。在人类所占比例较小的Proteobacteria 和 Actinobacteria在倭蜂猴中更为普遍,而所占比例较大的Firmicutes在倭蜂猴中所占的比例却较小。同时,通过对倭蜂猴与人及其他一些动物的胃肠道宏基因组数据进行双向聚类发现,虽然倭蜂猴和小鼠肠道系统的功能有差异,但其微生物群落结构却最为相似[73]。
3.2 筛选功能活性物质
动物胃肠道中栖息着大量的微生物,能分泌纤维素酶、木聚糖酶、淀粉酶和酯酶等水解酶,在动物消化过程中起着积极的不可忽视的作用。然而,由于胃肠道微生物大都是专性厌氧菌,可培养性很低,因此传统的微生物纯培养技术仅能分离获得胃肠道中约 15%~20%的微生物[70,74],即使在最近的研究中利用全面的厌氧培养和高通量16S测序技术,也只有56%的胃肠道微生物得以培养[75]。因此,还有近一半的胃肠道微生物因为无法培养而难以鉴定,同时,这也大大限制了通过分离培养微生物来发现和筛选新基因及生物活性物质的广泛性和有效性。
宏基因组学避开了微生物分离培养的问题,极大地扩展了微生物资源的利用空间,为寻找和发现新的功能基因及生物催化剂提供了新的研究策略。目前,国内外研究者已通过构建动物胃肠道微生物宏基因组文库或对宏基因组进行直接测序,成功筛选到多种糖苷水解酶及淀粉酶、脂酶/酯酶等酶类基因 (表5)。由于植食性动物胃肠道是一个植物纤维素剧烈降解的环境,存在其中的微生物具备了相应的特殊生理、代谢特点,能产生各种纤维素分解酶类,因此,近年来利用宏基因组学技术从动物胃肠道筛选酶基因的研究主要集中在植食性动物。对白蚁[80]、牛[86,90]、袋鼠[87]和大熊猫[63]胃肠道微生物宏基因组的研究发现,动物胃肠道中含有种类和数量丰富的植物生物质降解酶类。Hess等[90]通过分析牛瘤胃 2 791个经证实具有生物质降解酶功能的基因同源性发现,只有1%与现有数据库中的基因有较高的相似性。此外,不同植食性动物由于摄食种类不同,其胃肠道内的植物生物质降解酶种类也存在很大差异。Warnecke等[80]通过对白蚁肠道微生物宏基因组的研究发现,其木质纤维素的降解以 GH5、GH94、GH51、GH8、GH9、GH44、GH45 和GH74的纤维素酶为主,然而却缺乏多数微生物纤维素酶系中的重要组成成分GH6和 GH48。上述研究表明,植食性动物胃肠道内蕴涵着大量潜在的纤维素分解酶类基因资源,为新型纤维素分解酶类的研究开发及其在生物能源领域的应用提供了丰富的资源。
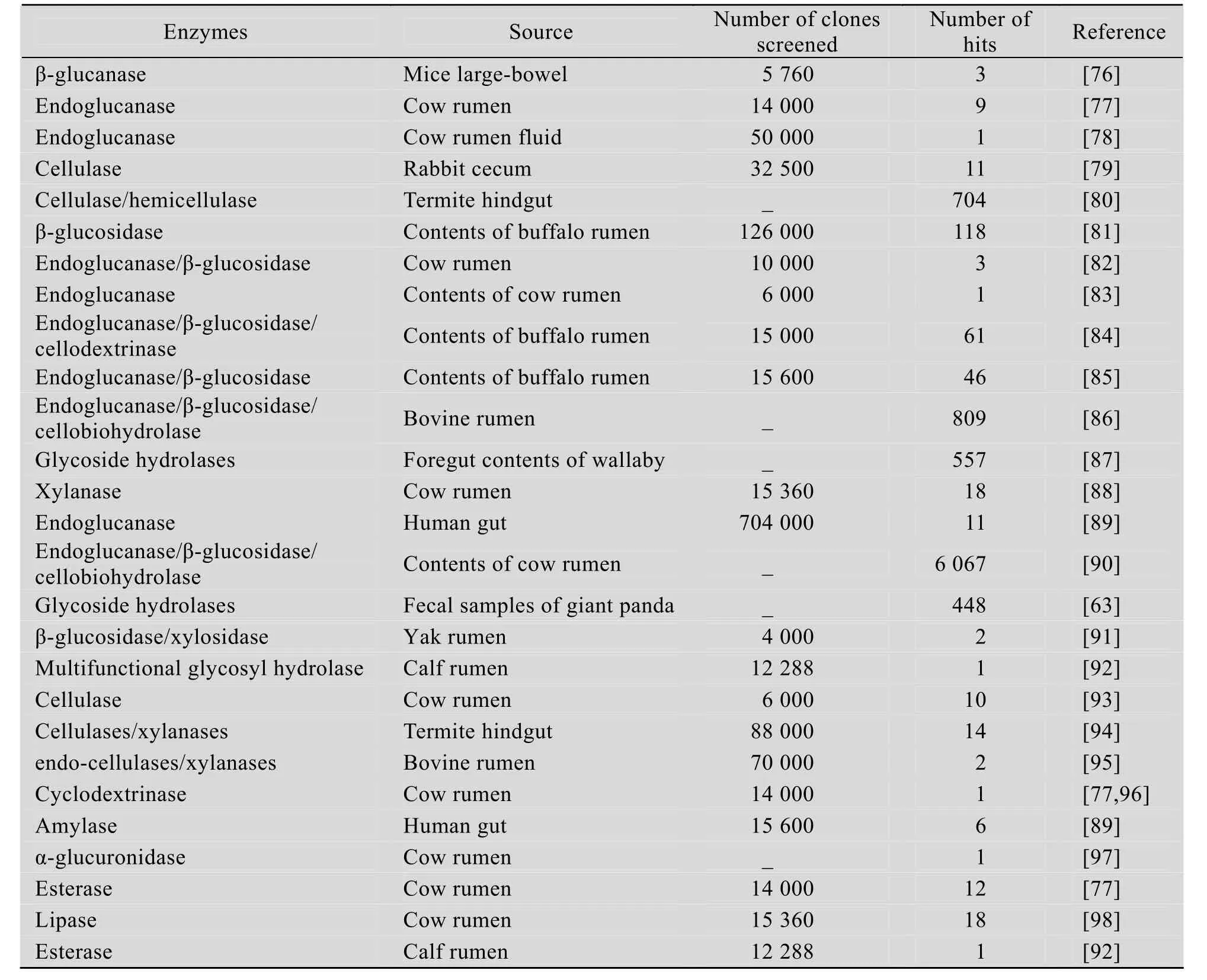
表5 从人和动物胃肠道宏基因组中筛选到的酶类Table 5 Identified enzymes from metagenome of animal gastrointestinal tract
本实验室在对低等灵长类动物倭蜂猴进行研究时,构建了Fosmid宏基因组文库,并利用功能筛选法从50 000个克隆中筛选到8个产淀粉酶的阳性克隆,通过亚克隆获得一个新的淀粉酶基因,并对其进行了生化鉴定(结果待发表),从而也证实了利用功能筛选从胃肠道微生物宏基因组中发现和筛选新基因及生物活性物质的可行性。
3.3 研究胃肠道微生物与人类健康和疾病的关系
人体含有的细菌细胞数量是人细胞的10倍多,其中大多数细菌细胞位于人肠道中,这些肠道菌群如何影响我们的健康?最近的宏基因组学研究表明,胃肠道微生物组的异常与人类的多种疾病过程有关联。
3.3.1 胃肠道微生物与肥胖
全球范围的肥胖问题促使研究者更加努力地寻找影响能量平衡的环境因素和人类自身因素,宏基因组为肠道微生物在肥胖形成中所起的作用提供了重要证据。Ley等[99]的研究发现,肥胖老鼠与同窝出生的瘦老鼠相比,盲肠内容物中Bacteroidetes和Firmicutes的比例发生明显变化。然而这一结果并不能证明是Bacteroidetes菌群的变化导致了体重的增加,还是由于特殊的饮食摄入所致。然而,Turnbaugh等通过宏基因组和生化分析所进行的研究证实[100],Bacteroidetes和Firmicutes细菌相对丰度的变化影响着肠道微生物的代谢能力,肥胖小鼠的肠道微生物具有更高的从饮食中摄取能量的能力;与定植有“瘦型微生物组”的小鼠相比,“肥胖型微生物组”可以促使小鼠体重的增加,从而说明肥胖这一表型可以通过微生物组传递。
为阐明宿主基因型、环境暴露以及肥胖对肠道微生物的影响,Turnbaugh等[101]利用宏基因组学技术,对 154个肥胖或瘦表型一致的同卵双生和异卵双生成年女性及其母亲的粪便微生物群落进行了研究。结果表明,与瘦者相比肥胖者体内拟杆菌门细菌比例降低,而放线菌门细菌比例升高;肥胖者肠道微生物多样性降低,助长了异常的能量输入,并且微生物处理碳水化合物的磷酸转移酶系统较瘦型志愿者更为富集。可见,微生物组成在门水平上的变化、细菌多样性的降低以及代谢途径改变均与肥胖相关。
3.3.2 胃肠道微生物与糖尿病
肥胖是Ⅱ型糖尿病发生的主要危险因素之一,最近研究者开始推测肠道菌群除影响肥胖外,可能对糖尿病也会产生影响。Qin等研究了来自中国的 345人的肠道细菌宏基因组,其中171人患有Ⅱ型糖尿病,结果表明人体内肠道细菌的组成在Ⅱ型糖尿病的发展过程中发挥重要作用[102]。此外,Brown等通过分析4例自体免疫I型糖尿病个案的粪便微生物宏基因组文库,认为自体免疫性疾病包括 I型糖尿病的产生与体内细菌的群落结构有关[103]。
3.3.3 胃肠道微生物与炎症性肠病(Inflammatory bowel disease, IBD)
Manichanh等[104]通过构建宏基因组文库对6个健康人和6个CD (Crohn’s disease)患者的粪便微生物组进行了研究。在所筛选的1 190个克隆中,鉴定出 125个非冗余核糖体基因型,主要为拟杆菌门和厚壁菌门。其中,在健康人体肠道中鉴定出 43种厚壁菌门细菌,而在CD患者中仅鉴定出 13种,尤其是柔嫩梭菌在CD患者中的比例显著低于正常人,可见,细菌组成多样性的降低是CD患者粪便微生物的特征。
作为MetaHIT计划的一部分,2010年,Qin等[2]比较分析了 IBD患者与健康人的粪便微生物宏基因组,发现IBD患者比健康人的肠道菌群总基因平均少25%,这一研究同样显示IBD患者与健康人相比具有较低的细菌多样性。
3.3.4 胃肠道微生物与其他疾病
Chen等[105]通过对16S rRNA V3区扩增子进行焦磷酸测序,分析了36个肝硬化病人和24个正常人肠道菌群结构的变化。研究发现,门水平上肝硬化患者肠道中拟杆菌门细菌的比例显著降低,变形菌门和梭杆菌门细菌显著富集,科水平上肝硬化患者肠道中则显著富集肠杆菌科、韦荣球菌科和链球菌科的细菌。
此外,最新的研究还证实人肠道菌群变化与表现出症状的动脉粥样硬化和中风相关联,Karlsson等[106]对中风病人和健康人肠道微生物的宏基因组进行比较发现,其肠道菌群存在明显差异,且病人的宏基因组中富含编码肽聚糖生物合成的基因,而健康对照组中富含编码八氢番茄红素脱氢酶的基因。
4 展望
宏基因组学的应用大大拓宽了人和动物胃肠道微生物的研究内容和范围,使单纯的微生物群落结构研究逐步过渡到功能代谢研究。然而,要想深入了解胃肠道生态系统中特定微生物的实际地位或作用,实现肠道菌群与宿主间的“交谈”,鉴定微生物在胃肠道生态环境中生存所需基因,研究胃肠道微生物和宿主免疫系统之间的互作机制及胃肠道菌群调节宿主多种表型的机制至关重要。胃肠道微生物的组成对哺乳动物功能基因组及其健康和疾病的研究具有重要影响,因此,进一步深入了解人类及其他动物胃肠道微生物群落的组成和功能,对于监控、预防、干预肠道菌群,探索胃肠道疾病和其他系统性疾病的治疗方法,以及促进胃肠道微生物资源的开发和利用将具有重要意义。
[1]Nicholson JK, Holmes E, Wilson ID. Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol, 2005,3(5): 431−438.
[2]Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 2010, 464(7285): 59−65.
[3]Handelsman J, Rondon MR, Brady SF, et al.Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol, 1998, 5(10): 245−249.
[4]Culley AI, Lang AS, Suttle CA. Metagenomic analysis of coastal RNA virus communities. Science,2006, 312(5781): 1795–1798.
[5]Narasingarao P, Podell S, Ugalde JA, et al. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J, 2012, 6(1): 81–93.
[6]Pignatelli M, Moya A. Evaluating the fidelity of de novo short read metagenomic assembly using simulated data. PLoS ONE, 2011, 6(5): e19984.
[7]Mende DR, Waller AS, Sunagawa S, et al.Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE, 2012, 7(2): e31386.
[8]The Human Microbiome Project Consortium. A framework for human microbiome research. Nature,2012, 486(7402): 215–221.
[9]Batzoglou S, Jaffe D, Stanley K, et al. ARACHNE:A whole-genome shotgun assembler. Genome Res,2002, 12(1): 177−189.
[10]Huang X, Wang J, Aluru S, et al. PCAP: a whole-genome assembly program. Genome Res,2003, 13(9): 2164−2170.
[11]Miller JR, Delcher AL, Koren S, et al. Aggressive assembly of pyrosequencing reads with mates.Bioinfomaticts, 2008, 24(24): 2818−2824.
[12]Lai B, Ding R, Li Y, et al. A de novo metagenomic assembly program for shotgun DNA reads.Bioinformatics, 2012, 28(11): 1455−1462.
[13]Warren RL, Sutton GG, Jones SJ, et al. Assembling millions of short DNA sequences using SSAKE.Bioinformatics, 2007, 23(4): 500−501.
[14]Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs.Genome Res, 2008, 18(5): 821−829.
[15]Butler J, MacCallum I, Kleber M, et al. ALLPATHS:de novo assembly of whole-genome shotgun microreads. Genome Res, 2008, 18(5): 810−820.
[16]Simpson JT, Wong K, Jackman SD, et al. ABySS: a parallel assembler for short read sequence data.Genome Res, 2009, 19(6): 1117−1123.
[17]Boisvert S, Laviolette F, Corbeil J. Ray:simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol, 2010, 17(11): 1519−1533.
[18]Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res, 2010, 20(2): 265−272.
[19]Laserson J, Jojic V, Koller D. Genovo: de novo assembly for metagenomes. J Comput Biol, 2011,18(3): 429−443.
[20]Treangen TJ, Sommer DD, Angly FE, et al. Next generation sequence assembly with AMOS. Curr Protoc Bioinformatics, 2011, 33: 11.8.1–11.8.18.
[21]Peng Y, Leung HC, Yiu SM, et al. Meta-IDBA: a de novo assembler for metagenomic data.Bioinformatics, 2011, 27(13): i94−101.
[22]Namiki T, Hachiya T, Tanaka H, et al. MetaVelvet:an extension of Velvet assembler to de novo metagenome assembly from short sequence reads.Nucleic Acids Res, 2012, 40(20): e155.
[23]Pell J, Hintze A, Canino-Koning R, et al. Scaling metagenome sequence assembly with probabilistic de Bruijn graphs. Proc Natl Acad Sci USA, 2012,109(33): 13272–13277.
[24]Peng Y, Leung HC, Yiu SM, et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth.Bioinformatics, 2012, 28(11): 1420−1428.
[25]Kultima JR, Sunagawa S, Li J, et al. MOCAT: a metagenomics assembly and gene prediction toolkit.PLoS ONE, 2012, 7(10): e47656.
[26]Yok NG, Rosen GL. Combining gene prediction methods to improve metagenomic gene annotation.BMC Bioinformatics, 2011, 12: 20.
[27]Segata N, Boernigen D, Tickle TL, et al.Computational meta'omics for microbial community studies. Mol Syst Biol, 2013, 9: 666.
[28]Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res, 2006, 34(19):5623−5630.
[29]Noguchi H, Taniguchi T, Itoh T.MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res, 2008, 15(6): 387−396.
[30]Hoff KJ, Lingner T, Meinicke P, et al. Orphelia:predicting genes in metagenomic sequencing reads.Nucleic Acids Res, 2009, 37:W101−105.
[31]Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res, 2010, 38(12): e132.
[32]Rho M, Tang H, Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res, 2010, 38(20): e191.
[33]Kelley DR, Liu B, Delcher AL, et al. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res, 2012, 40(1): e9.
[34]DeSantis TZ, Hugenholtz P, Larsen N, et al.Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol, 2006, 72(7): 5069−5072.
[35]Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol, 2007, 73(16): 5261−5267.
[36]Monzoorul Haque M, Ghosh TS, Komanduri D, et al.SOrt-ITEMS: sequence orthology based approach for improved taxonomic estimation of metagenomic sequences. Bioinformatics, 2009, 25(14):1722−1730.
[37]Schloss PD, Westcott SL, Ryabin T, et al.Introducing mothur: open-source,platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol, 2009, 75(23):7537−7541.
[38]Diaz NN, Krause L, Goesmann A, et al. TACOA:taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics, 2009, 10: 56.
[39]Schreiber F, Gumrich P, Daniel R, et al. Treephyler:fast taxonomic profiling of metagenomes.Bioinformatics, 2010, 26(7): 960−961.
[40]Caporaso JG, Kuczynski J, Stombaugh J, et al.QIIME allows analysis of high-throughput community sequencing data. Nat Methods, 2010,7(5): 335−336.
[41]Weber M, Teeling H, Huang S, et al. Practical application of self-organizing maps to interrelate biodiversity and functional data in NGS-based metagenomics. ISME J, 2010, 5(5): 918–928.
[42]Liu B, Gibbons T, Ghodsi M, et al. Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC Genomics, 2011, 12(Suppl 2): S4.
[43]Brady A, Salzberg S. PhymmBL expanded:confidence scores, custom databases, parallelization and more. Nat Methods, 2011, 8(5): 367.
[44]Parks DH, MacDonald NJ, Beiko RG. Classifying short genomic fragments from novel lineages using composition and homology. BMC Bioinformatics,2011, 12: 328.
[45]Jia P, Xuan L, Liu L, et al. MetaBinG: using GPUs to accelerate metagenomic sequence classification.PLoS ONE, 2011, 6(11): e25353.
[46]Rosen GL, Reichenberger ER, Rosenfeld AM. NBC:the Naive bayes classification tool webserver for taxonomic classification of metagenomic reads.Bioinformatics, 2011, 27(1): 127−129.
[47]Gerlach W, Stoye J. Taxonomic classification of metagenomic shotgun sequences with CARMA3.Nucleic Acids Res, 2011, 39(14): e91.
[48]Huson DH, Mitra S, Ruscheweyh HJ, et al. Integrative analysis of environmental sequences using MEGAN4.Genome Res, 2011, 21(9): 1552−1560.
[49]Patil KR, Roune L, McHardy AC. The PhyloPythiaS web server for taxonomic assignment of metagenome sequences. PLoS ONE, 2012, 7(6): e38581.
[50]Su X, Xu J, Ning K. Parallel-META: efficient metagenomic data analysis based on high-performance computation. BMC Syst Biol,2012, 6(Suppl 1): S16.
[51]Wu M, Scott AJ. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2.Bioinformatics, 2012, 28(7): 1033–1034.
[52]Kotera M, Hirakawa M, Tokimatsu T, et al. The KEGG databases and tools facilitating omics analysis: latest developments involving human diseases and pharmaceuticals. Methods Mol Biol,2012, 802: 19–39.
[53]Cantarel BL, Coutinho PM, Rancurel C, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res, 2009, 37: D233−238.
[54]Meyer F, Paarmann D, D'Souza M, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 2008, 9: 386.
[55]Markowitz VM, Chen IM, Chu K, et al. IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res,2012, 40: D123−129.
[56]Wu S, Zhu Z, Fu L, et al. WebMGA: a customizable web server for fast metagenomic sequence analysis.BMC Genomics, 2011, 12: 444.
[57]Li W. Analysis and comparison of very large metagenomes with fast clustering and functional annotation. BMC Bioinformatics, 2009, 10: 359.
[58]Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics, 2010, 26(6): 715−721.
[59]Goll J, Rusch DB, Tanenbaum DM, et al. METAREP:JCVI metagenomics reports--an open source tool for high-performance comparative metagenomics.Bioinformatics, 2010, 26(20): 2631−2632.
[60]Lingner T, Asshauer KP, Schreiber F, et al.CoMet--a web server for comparative functional profiling of metagenomes. Nucleic Acids Res, 2011,39: W518−523.
[61]Arndt D, Xia J, Liu Y, et al. METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res, 2012, 40:W88−95.
[62]Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science,2006, 312(5778): 1355–1359.
[63]Zhu L, Wu Q, Dai J, et al. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci USA, 2011, 108 (43): 17714−17719.
[64]Kurokawa K, Itoh T, Kuwahara T, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res, 2007,14(4): 169–181.
[65]Qu A, Brulc JM, Wilson MK, et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE, 2008, 3(8): e2945.
[66]Swanson KS, Dowd SE, Suchodolski JS, et al.Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J, 2011, 5(4): 639–649.
[67]Lamendella R, Domingo JW, Ghosh S, et al.Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol,2011, 11: 103.
[68]Tun HM, Brar MS, Khin N, et al. Gene-centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. J Microbiol Methods, 2012, 88(3): 369–376.
[69]Singh KM, Ahir VB, Tripathi AK, et al.Metagenomic analysis of Surti buffalo (Bubalus bubalis)rumen: a preliminary study. Mol Biol Rep,2012, 39(4): 4841−4848.
[70]Li RW, Connor EE, Li C, et al. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol, 2012, 14(1):129−139.
[71]Pope PB, Mackenzie AK, Gregor I, et al.Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS ONE, 2012, 7(6): e38571.
[72]Bhatt VD, Dande SS, Patil NV, et al. Molecular analysis of the bacterial microbiome in the forestomach fluid from the dromedary camel(Camelus dromedarius). Mol Biol Rep, 2013, 40(4):3363−3371.
[73]Xu B, Xu W, Yang F, et al. Metagenomic analysis of the pygmy loris fecal microbiome reveals unique functional capacity related to metabolism of aromatic compounds. PLoS ONE, 2013, 8(2): e56565.
[74]Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science,2005, 308(5728): 1635–1638.
[75]Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice.Proc Natl Acad Sci USA, 2011, 108(15): 6252–6257.
[76]Walter J, Mangold M, Tannock GW. Construction,analysis, and beta-glucanase screening of a bacterial artificial chromosome library from the large-bowel microbiota of mice. Appl Environ Microbiol, 2005,71(5): 2347−2354.
[77]Ferrer M, Golyshina OV, Chernikova TN, et al.Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora.Environ Microbiol, 2005, 7(12): 1996−2010.
[78]Palackal N, Lyon CS, Zaidi S, et al. A multifunctional hybrid glycosyl hydrolase discovered in an uncultured microbial consortium from ruminant gut. Appl Microbiol Biotechnol, 2007, 74(1):113−124.
[79]Feng Y, Duan CJ, Pang H, et al. Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol, 2007, 75(2): 319−328.
[80]Warnecke F, Luginbuhl P, Ivanova N, et al.Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature,2007, 450(7169): 560−565.
[81]Guo H, Feng Y, Mo XC, et al. Cloning and expression of a β-glucosidase gene umcel3G from metagenome of buffalo rumen and characterization of the translated product. Chin J Biotech, 2008, 24(2):232−238 (in Chinese).郭鸿, 封毅, 莫新春, 等. 水牛瘤胃宏基因组的一个新的 β-葡萄糖苷酶基因 umcel3G的克隆、表达及其表达产物的酶学特性. 生物工程学报, 2008,24(2): 232−238.
[82]Wang F, Li F, Chen G, et al. Isolation and characterization of novel cellulase genes from uncultured microorganisms in different environmental niches. Microbiol Res, 2009, 164(6):650−657.
[83]Shedova EN, Berezina OV, Lunina NA, et al.Cloning and characterization of a large metagenomic DNA fragment containing glycosyl-hydrolase genes.Mol Gen Mikrobiol Virusol, 2009, 24(1): 11−15.
[84]Duan CJ, Xian L, Zhao GC, et al. Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J Appl Microbiol, 2009, 107(1): 245−256.
[85]Liu L, Feng Y, Duan CJ, et al. Isolation of a gene encoding endoglucanase activity from uncultured microorganisms in buffalo rumen. World J Microb Biot, 2009, 25(6): 1035−1042.
[86]Brulc JM, Antonopoulos DA, Miller ME, et al.Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci USA, 2009,106(6): 1948−1953.
[87]Pope P, Denman S, Jones M, et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci USA, 2010, 107(33):14793−14798.
[88]Zhao S, Wang J, Bu D, et al. Novel glycoside hydrolases identified by screening a Chinese Holstein dairy cow rumen-derived metagenome library. Appl Environ Microbiol, 2010, 76(19):6701–6705.
[89]Tasse L, Bercovici J, Pizzut-Serin S, et al.Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes.Genome Res, 2010, 20(11): 1605–1612.
[90]Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science, 2011, 331(6016):463−467.
[91]Bao L, Huang Q, Chang L, et al. Cloning and characterization of two β-glucosidase/xylosidase enzymes from yak rumen metagenome. Appl Biochem Biotechnol, 2012, 166(1): 72−86.
[92]Ferrer M, Ghazi A, Beloqui A, et al. Functional metagenomics unveils a multifunctional glycosyl hydrolase from the family 43 catalysing the breakdown of plant polymers in the calf rumen.PLoS ONE, 2012, 7(6): e38134.
[93]Gong X, Gruninger RJ, Qi M, et al. Cloning and identification of novel hydrolase genes from a dairy cow rumen metagenomic library and characterization of a cellulase gene. BMC Res Notes, 2012, 5: 566.
[94]Nimchua T, Thongaram T, Uengwetwanit T, et al.Metagenomic analysis of novel lignocellulose-degrading enzymes from higher termite guts inhabiting microbes. J Microbiol Biotechnol, 2012, 22(4): 462−469.
[95]Rashamuse KJ, Visser DF, Hennessy F, et al.Characterisation of two bifunctional cellulase-xylanase enzymes isolated from a bovine rumen metagenome library. Curr Microbiol, 2013, 66(2): 145−151.
[96]Ferrer M, Beloqui A, Golyshina OV, et al.Biochemical and structural features of a novel cyclodextrinase from cow rumen metagenome.Biotechnol J, 2007, 2(2): 207–213.
[97]Lee CC, Kibblewhite RE, Wagschal K, et al.Isolation of α-glucuronidase enzyme from a rumen metagenomic library. Protein J, 2012, 31(3):206–211.
[98]Zhao SG, Wang JQ, Liu KL, et al. Screening and characterization of lipase from a metagenome library of dairy rumen microflora. Chin J Biotech, 2009,25(6): 869−874 (in Chinese).赵圣国, 王加启, 刘开朗, 等. 奶牛瘤胃微生物元基因组文库中脂肪酶的筛选与酶学性质. 生物工程学报, 2009, 25(6): 869−874.
[99]Ley RE, Bäckhed F, Turnbaugh P, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA, 2005, 102(31): 11070−11075.
[100]Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature, 2006, 444(7122):1027–1031.
[101]Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature,2009, 457(7228): 480–484.
[102]Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes.Nature, 2012, 490(7418): 55−60.
[103]Brown CT, Davis-Richardson AG, Giongo A, et al.Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE, 2011,6(10): e25792.
[104]Manichanh C, Rigottier-Gois L, Bonnaud E, et al.Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut,2006, 55(2): 205–211.
[105]Chen Y, Yang F, Lu H, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology, 2011, 54(2): 562–572.
[106]Karlsson FH, Fåk F, Nookaew I, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun, 2012, 3: 1245.
