首例合并眼球震颤的X连锁网状色素异常症的家系分析
2013-11-01戴珊赵暕宋清华
戴珊,赵暕,宋清华
(北京大学第三医院,北京100191)
X连锁网状色素异常症(X-linked reticulate pigmentary disorder with systemic manifestations in males,PDR)是一种极为罕见的遗传性色素异常性疾病,遗传模式为X染色体遗传,主要为男性发病,表现为出生不久后出现全身弥漫分布的大片色素沉着,其上可出现多发点状或者圆形的大小不等色素减退斑,可融合形成网状改变,伴有额部及双鬓毛发呈向上后侧弯曲。此外,患者还可以合并畏光、角膜营养不良,少汗、反复发作性呼吸道感染、胃肠道炎症、尿道狭窄,身材矮小等症状[1]。目前国外仅报道6个家系[1-3]1个散发病例[4],国内仅报道2例散发病例[5-6]。我们报道首例合并眼球震颤的患者及其家系,结合文献,分析遗传性眼球震颤的致病基因,为定位本病的致病基因提供新的方向。
1 资料与方法
患者男,44岁,2岁时无明显诱因于颈部,躯干、四肢、手足出现泛发性褐色至深褐色斑疹,针尖至绿豆大小,颜色深浅不一,大小不等,形状不规则,相互融合成网状,皮损区皮肤质软、光滑,表面无鳞屑、皮肤萎缩、毛细血管扩张,掌跖及指(趾)甲无改变,斑疹间可见正常皮肤,无自觉症状,皮损与日晒无关,不随季节发生变化,见图1,头发粗糙、干燥,额部及双侧鬓角头发向上后侧弯曲,同时发现有双眼眼球震颤。体格与智力发育正常,营养良好,牙齿无异常,眼睛无畏光,口腔黏膜及肛周黏膜未见色素脱失斑。眼科检查示双眼水平性眼球震颤伴外斜视,角膜,眼底检查无异常。系统检查未见其他异常。平素体健,无少汗、反复肺炎、胃肠道炎症及排尿困难等病史。实验室及辅助检查:血、尿、粪常规及生化检查均正常。激素节律检查及促肾上腺皮质激素水平均正常。胸片无异常。父母非近亲结婚,家族中有同样病史,所有患者均为男性,家族中所有女性均正常,有隔代遗传现象,符合X连锁隐性遗传模式,见图2。于患者腹部典型色素沉着斑处切取皮肤组织,分别进行组织化学病理检查及电镜检查。
2 结果
2.1 皮损组织病理检查 表皮大致正常,基底层黑素细胞数量正常,部分区域黑素颗粒增多;真皮浅层血管周围可见轻度淋巴细胞为主的炎细胞浸润,未见淀粉样物质沉积,刚果红染色阴性,见图3。
2.2 电镜检查 基底层黑素细胞及黑素体数量及结构均正常,黑素颗粒增多,见图4。
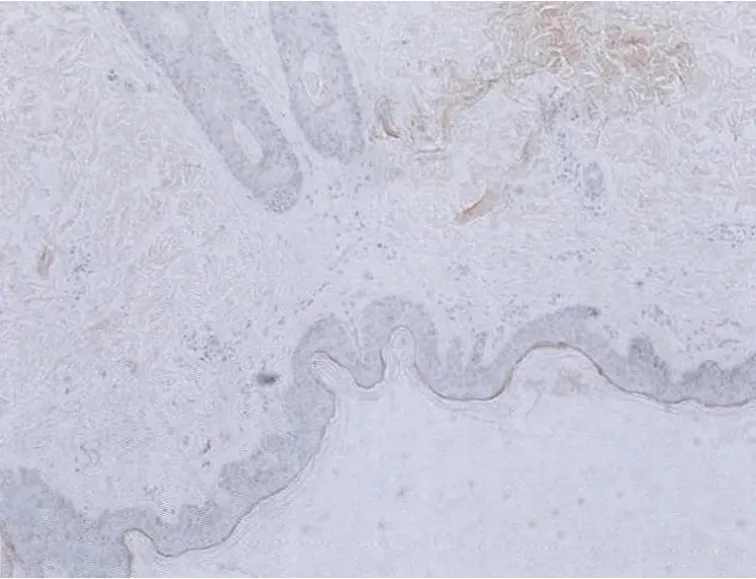
图3 基底层可见较多黑素颗粒,刚果红染色阴性(HE及刚果红染色×100)
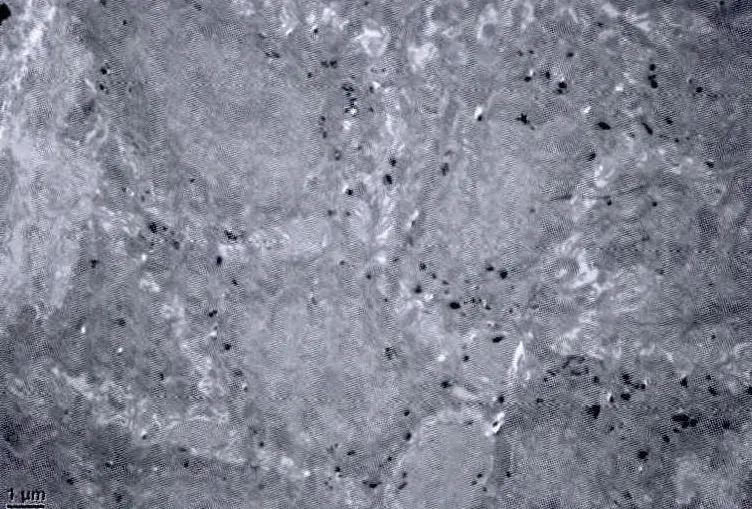
图4 基底层黑素颗粒增多,黑素细胞及黑素体数量及结构均正常(醋酸双氧铀染色×5000)
3 讨论
PDR是一种非常罕见的遗传性色素异常性疾病,于1981年由Partington等[7]首次报道一个加拿大家系,目前国外仅报道6个家系及一个散发病例,国内仅报道2例散发病例,尚无家系报道。该病遗传模式为X染色体遗传,主要为男性发病。病人出生时即可表现出弥漫的网状色素沉着及色素减退,伴有少汗、严重的新生儿肠炎、反复发作的肺炎、角膜营养不良、畏光、眼睑痉挛、胃肠道炎症、尿道狭窄、生长发育迟缓以及特征性的毛发改变,如头发粗糙,额部及双鬓毛发呈向上后侧弯曲。女性携带者由于X染色体不完全失活可表现正常,或仅表现为沿着Blaschko线分布的片状色素沉着斑,而无其他系统受累表现。该病的致病基因目前被定位于Xp22.11-Xp21.3的一段约4.9 cm区域中[8]。
不同性别的患者皮肤组织病理学表现相同,但无诊断特异性。均为基底层黑素颗粒增加,可有轻度角化亢进及角化不全,色素失禁,真皮内噬黑素细胞形成等。电镜下可表现为基底层色素增加,黑素体、黑素细胞及角质形成细胞形态结构均正常[9]。斑块状淀粉样物质在皮肤组织中的沉积目前仅见于首次报道的加拿大家系的病人中,且不伴有其他系统组织淀粉样变[7]。组织化学检查及刚果红染色均未证明我们的病人皮损中有淀粉样物质沉积。Partington等[9]推测淀粉样物质的沉积是由于病人角质形成细胞变性,与本病发生机制无关。
我们报道的该患者皮损具有典型的网状色素异常改变,出生不久即发病,伴有皮肤外系统受累表现即眼球震颤,皮损组织病理及电镜检查示基底层黑素颗粒增多,黑素细胞数量及结构正常,无淀粉样蛋白沉积,诊断符合X连锁网状色素异常症。具有典型家系,符合X连锁隐性遗传模式,这在国内尚属首次报道。同时,我们对其皮肤组织进行了常规组织化学检查,刚果红染色及电镜检查,进一步从病理学上支持了该诊断,验证了既往报道的结果,也填补了国内对该病皮损电镜检查结果的空白。
该患者及其家系中其他患者无少汗、反复发作肺炎或胃肠道炎症等既往报道的常见合并症,而均单纯表现为眼球震颤,这在国内外也尚未见相关报道。国内报道的较少见合并症为耳廓软骨发育不良[5],国外曾报道合并小耳畸形[3],正细胞正色素性贫血,掌跖皮肤角化,杵状指(趾),指骨短缩发育不良[7],并指[8]。通过查阅文献我们发现,眼球震颤可为眼白化病的一种变异表现,以发作性有节律的双侧眼球摆动为典型症状,可伴有视力下降,弱视,虹膜透明化,黄斑发育不良以及眼底脉络膜色素减退,而皮肤及毛发色素正常[10]。多在婴儿期起病。其致病基因定位于Xp22.3的GPR143,整个基因编码404个氨基酸,其蛋白产物OA1是一种色素细胞特异的膜糖蛋白(60 kDa,非糖基化前体为45~48 ku的双联体),具有高度保守性。OA1表达于眼和表皮的黑素细胞内的黑素体膜,调节黑色素的发生和成熟,在黑素颗粒的生物形成过程中起重要作用[11-12]。本病患者眼科检查未见色素膜及黄斑异常,色素异常单纯表现在皮肤中,可除外眼白化病可能。而本病与眼白化病同为色素异常性疾病,两者的发生可能与不同部位黑素形成异常相关,可以一定程度上解释本病与眼白化病的变异型,即眼球震颤同时出现。且GPR143恰好位于本病致病基因既往被定位的区段中,我们假设该基因不同部位的突变很可能在引起眼球震颤的同时导致皮肤网状色素异常。这也对今后进一步定位X连锁网状色素异常症的致病基因提供了一个高度可能的相关位点。
[1]Jaeckle Santos LJ,Xing C,Barnes RB,et al.Refined mapping of X-linked reticulate pigmentary disorder and sequencing of candidate genes[J].Hum Genet,2008,123:469-476.
[2]Fernandez-Guarino M,Torrelo A,Fernandez-Lorente M,et al.X-linked reticulate pigmentary disorder:report of a new family[J].Eur J Dermatol,2008,18:102-103.
[3]Fraile G,Norman F,Reguero ME,et al.Cryptogenic multifocal ulcerous stenosing enteritis(CMUSE)in a man with a diagnosis of X-linked reticulate pigmentary disorder (PDR)[J].Scand J Gastroenterol,2008,43:506-510.
[4]Kim BS,Seo SH,Jung HD,et al.X-Linked reticulate pigmentary disorder in a female patient[J].Int J Dermatol,2010,49:421-425.
[5]林志淼,杨勇.X连锁网状色素异常症1例[J].临床皮肤科杂志,2011,40(10):634-635.
[6]乌云,那顺布和,银花.X连锁网状色素异常症1例[J].中国麻风皮肤病杂志,2013,29(1):42-43.
[7]Partington MW,Marriott PJ,Prenticd RSA,et al.Familial cutaneous amyloidosis with systemic manifestations in males[J].Am J Med Genet 1981,10:65-75.
[8]Gedeon AK,Mulley JC,Kozman H,et al.Localization of the gene for X-linked reticulate pigmentary disorder with systemic manifestations(RPD),previously known as X-linked cutaneous amyloidosis[J].Am J Med Genet 1994,52:75-78.
[9]PartingtonMW,PrenticeRS.X-linked cutaneous amyloidosis:further clinical and pathological observations[J].Am J Med Genet,1989,32:115-119.
[10]Lorenz B,Gampe E.Analyse von 180 Patienten mit sensorischem Defektnystagmus(SDN)undkongenitalemidiopathischemNystagmus(CIN)[J].Klin Monatsbl Augenheilkd,2001,218:3-12.
[11]Cortese K,Giordano F,Surace EM,et al.The ocular albinism type 1(OA1)gene controls melanosome maturation and size[J].Invest Ophthalmol Vis Sci,2005,46:4358-4364.
[12]Schiaffino MV,d’Addio M,Alloni A,et al.Ocular albinism:evidence for a defect in an intracellular signal transduction system[J].Nat Genet,1999,23:108-112.
