硅烯对含氢氯硅烷选择性Si—X键插入反应的理论研究
2013-10-28杨雪敏李志芳邱化玉蒋剑雄来国桥
杨雪敏,金 娟,徐 征,李志芳,邱化玉,蒋剑雄,来国桥
(杭州师范大学有机硅化学及材料技术教育部重点实验室, 浙江 杭州 310012)
硅烯对含氢氯硅烷选择性Si—X键插入反应的理论研究
杨雪敏,金 娟,徐 征,李志芳,邱化玉,蒋剑雄,来国桥
(杭州师范大学有机硅化学及材料技术教育部重点实验室, 浙江 杭州 310012)
采用密度泛函B3LYP/6-31++G(d,p)算法,比较了通过位阻效应稳定的二烷基取代硅烯和通过电子效应稳定的二氨基取代硅烯对含氢氯硅烷Si—X(X=H或Cl)键插入反应的反应选择性差异.研究结果表明:(1) 二氨基取代硅烯的Si—X键插入反应的反应活性低且放热不明显,产物在热力学上远不及二烷基取代硅烯的产物稳定;(2) 二氨基取代硅烯对二甲基氢氯硅烷(Me2SiHCl)表现出了与二烷基取代硅烯不同的反应选择性,Si—Cl插入反应优先于Si—H插入反应.其根本原因在于Si—H插入反应过渡态能量的稳定性对硅烯的亲电性作用的依赖性显著大于Si—Cl插入反应.
理论计算;硅烯;含氢氯硅烷;Si—X插入反应
0 引 言
硅烯是有机硅化学中一种重要的反应中间体,有关它的反应特性及机理吸引了许多研究者的兴趣.σ键插入反应是硅烯的重要特性反应之一,在常规方法难以获得的新型有机硅化合物合成中具有重要的价值.自1971年首次发现了H2Si能插入Si—H键以来[1],在近半个世纪的时间里人们陆续发现它还能与其它许多σ键发生插入反应,包括 Si—N、Si—O、Si—Cl、Si—Si、具有环张力的C—O、C—H(分子内)、C—X(X=Cl, Br) 、N—H、N—M(M=Li, Na, K, Si(II), Ge(II), Sn(II), Pb(II)) 、O—H、Sn(II)—Cl, Sn(II)—C、Li—C、Li—Si、B—C等[2].
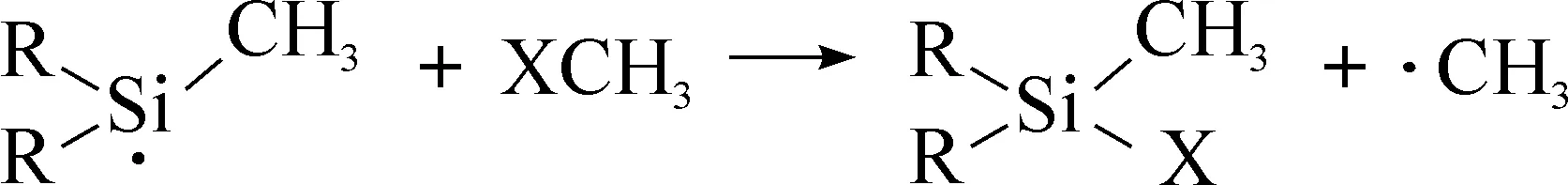
许多研究者从理论计算和实验角度出发对硅烯的部分插入反应的机理进行了研究,发现不同类型化学键其反应机理则不尽相同.对于C—X(X=卤素)键,硅烯是通过自由基机理实现键插入的,包括链引发(1)、链增长(2),(3),(4)等过程[3]:

(1)

(2)
(3)
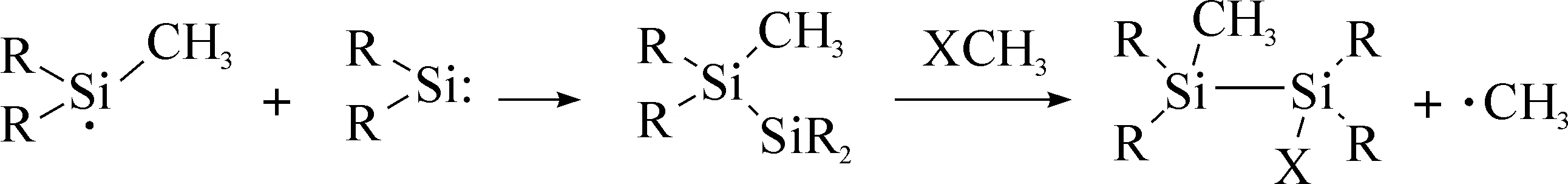
(4)
对于Si—H、N—H、O—H、Si—O和C—O等,硅烯则是通过协同机理实现键插入的[2,4],包括由这些化学键中电负性较强的原子与硅烯的p空轨道形成路易斯酸碱复合物和三元环的过渡态两个过程.

硅烯对Si—X键的插入反应被视为二硅烷重排反应[5]、多硅烷和聚硅烷的热解反应[6]和光解反应[7]的关键步骤.全面深入地了解这类反应的机理对于人们利用这类反应来高选择性地合成新型有机硅化合物具有重要意义.由于瞬态硅烯不易捕捉,难以获得有关反应机理的细节.人们往往通过各种室温稳定硅烯(图1)来了解硅烯各种特征反应的反应机理.目前人们已成功合成的稳定硅烯主要通过两种机制来稳定:一种是通过位阻效应从动力学角度避免硅烯二聚来实现稳定(1a),另一种则是通过电子效应从热力学角度实现稳定(1b-1e)[8].实验研究已发现由大位阻效应稳定的二烷基取代硅烯对二氯硅烷Si—X键表现出了高度的键插入反应选择性[9].同时,通过理论计算我们也已发现,硅烷中硅原子上的取代基具有显著的立体电子效应,对反应选择性起十分关键的作用[10].但目前有关通过电子效应稳定的硅烯对氢氯硅烷的反应选择如何仍为未知.本文将重点研究比较两类硅烯对氢氯硅烷Si—X键插入反应的反应选择性的差异,分析硅烯的电子结构对两种反应活化能的影响机制及规律.
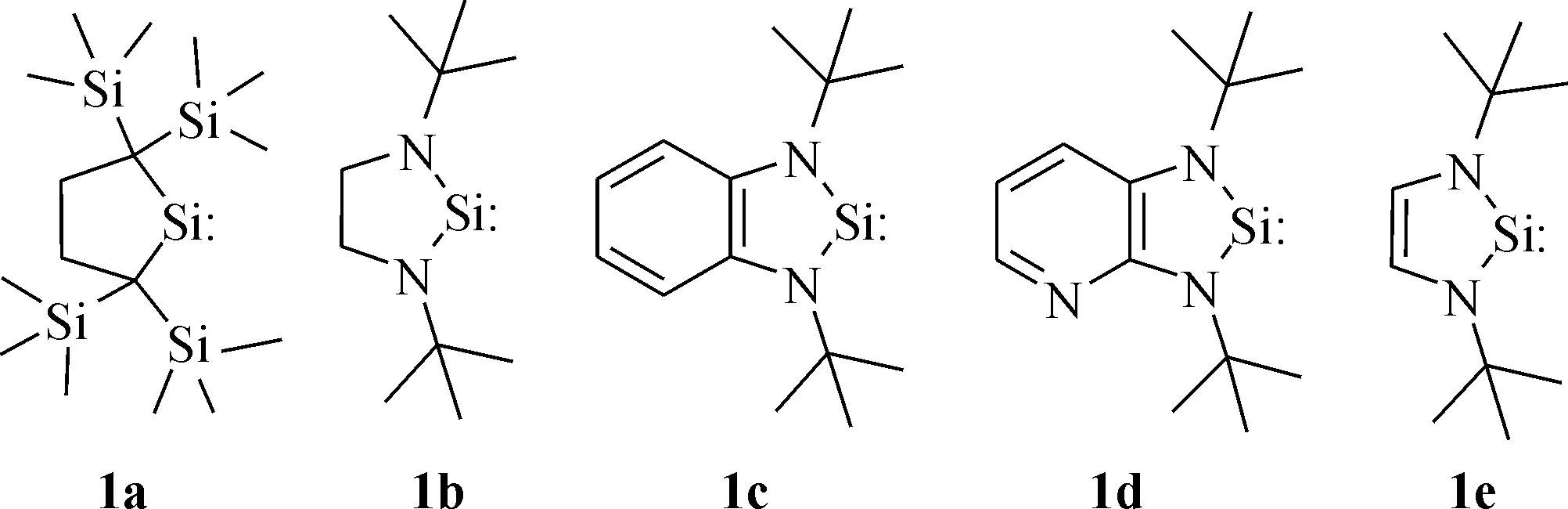

图1 室温稳定环状二烷基取代硅烯、二氨基取代硅烯和氢氯硅烷的模型分子Fig. 1 Model compounds for stable cyclic dialkylsilylene and diaminosilylene and hydrochlorosilane
1 模型分子与计算方法
1.1 模型分子的选择
为节约计算时间,选用1a′作为二烷基取代硅烯1a的模型分子,1b′和1c′作为具有不同共轭度的二氨基取代硅烯1b和1c的模型分子.选用四氢硅烷SiH4(2a)和三氢氯硅烷SiH3Cl(2b)作为Si—H键和Si—Cl插入反应的最简化模型分子,以此来对两类硅烯按式(5)进行σ键插入反应的活性进行比较.与此同时,选用二甲基氢氯硅烷2c来考察两类硅烯对同一分子内的两种键插入反应的反应选择性,选用二氢二氯硅烷(2d)来比较硅烷的电子效应对Si—Cl插入反应活化能的影响.
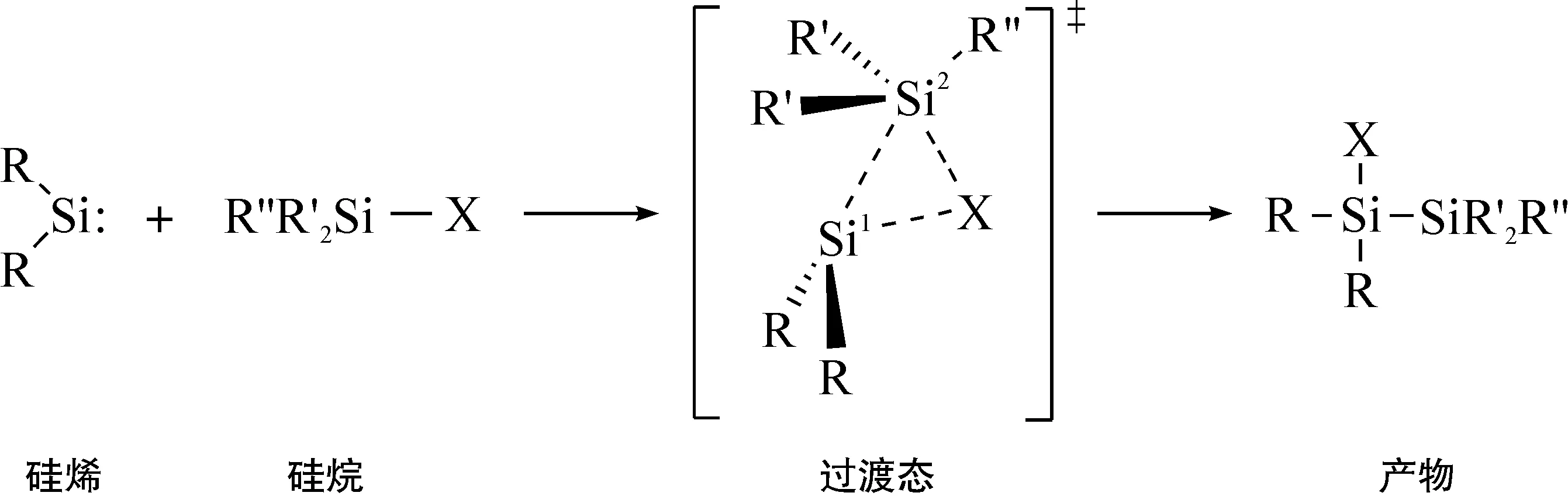
(5)
1. 2 计算方法
研究采用密度泛函理论方法, 在B3LYP/6-31++G(d,p)[11]水平下对反应势能面上所有驻点(反应物、复合物、过渡态和产物)进行了全参数几何结构优化,并通过同样算法下的频率分析确认各驻点的性质(稳定态无虚频、过渡态有一个虚频),通过内禀反应坐标(IRC)[12]计算确认过渡态与反应物或中间体和产物之间的相关性.文中所列各驻点的能量都进行了零点能(zero-point energy, ZPE) 校正,校正因子为1.所有工作在杭州师范大学有机硅实验室理论计算研究室的SGI/Altix 450服务器上采用Gaussian 03 量子化学计算软件完成[13].
2 结果与讨论
2.1 硅烯的取代基效应对Si—X键插入反应热力学的影响
表1列出了298.15 K计算得到的硅烯1a′-1c′与硅烷2a-2c进行Si—H和Si—Cl插入反应的反应焓变.从中可以看出二烷基取代硅烯1a′与2a的Si—H键插入、与2b的Si—Cl键插入都呈现显著的放热现象(35.0~37.8 kcal/mol),两种键插入反应产物在热力学上较反应物明显更稳定,且稳定能很接近.而二氨基取代硅烯1b′和1c′的放热量却显著减小(5.8~21.0 kcal/mol),且Si—Cl键插入反应的放热量比Si—H键插入反应大~10.0 kcal/mol.
2.2 硅烯的取代基效应对Si—X键插入反应动力学的影响
硅烯1a′-1c′与硅烷(2a-2d)两种键插入反应的过渡态的能量参数也列于表1中.B3LYP/6-31++G(d,p)水平上优化的硅烯1a′和1b′与硅烯2a和2b插入反应反应物、产物和过渡态的几何结构示于图2中.从表1中可以看出二氨基取代硅烯在反应动力学上也与二烷基取代硅烯具有很大的差别.二烷基取代硅烯1a′与2a的Si—H键插入、与2b的Si—Cl键插入反应的活化能分别为11.9 kcal/mol和10.9 kcal/mol,两种键插入反应的活化能差值仅为1 kcal/mol.而二氨基取代硅烯1b′和1c′参与上述类似反应的活化能则显著提高(20.5~40.7 kcal/mol),并且它们对2b的Si—Cl键和对2a的Si—H键插入的活化能差距也大幅度增加,Si—Cl键插入反应的活化能比Si—H键的低~15.0 kcal/mol.
2.3 硅烯对二甲基氢氯硅烷的选择性Si—X键插入反应及机理
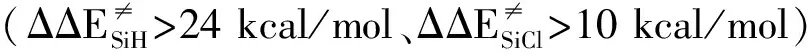
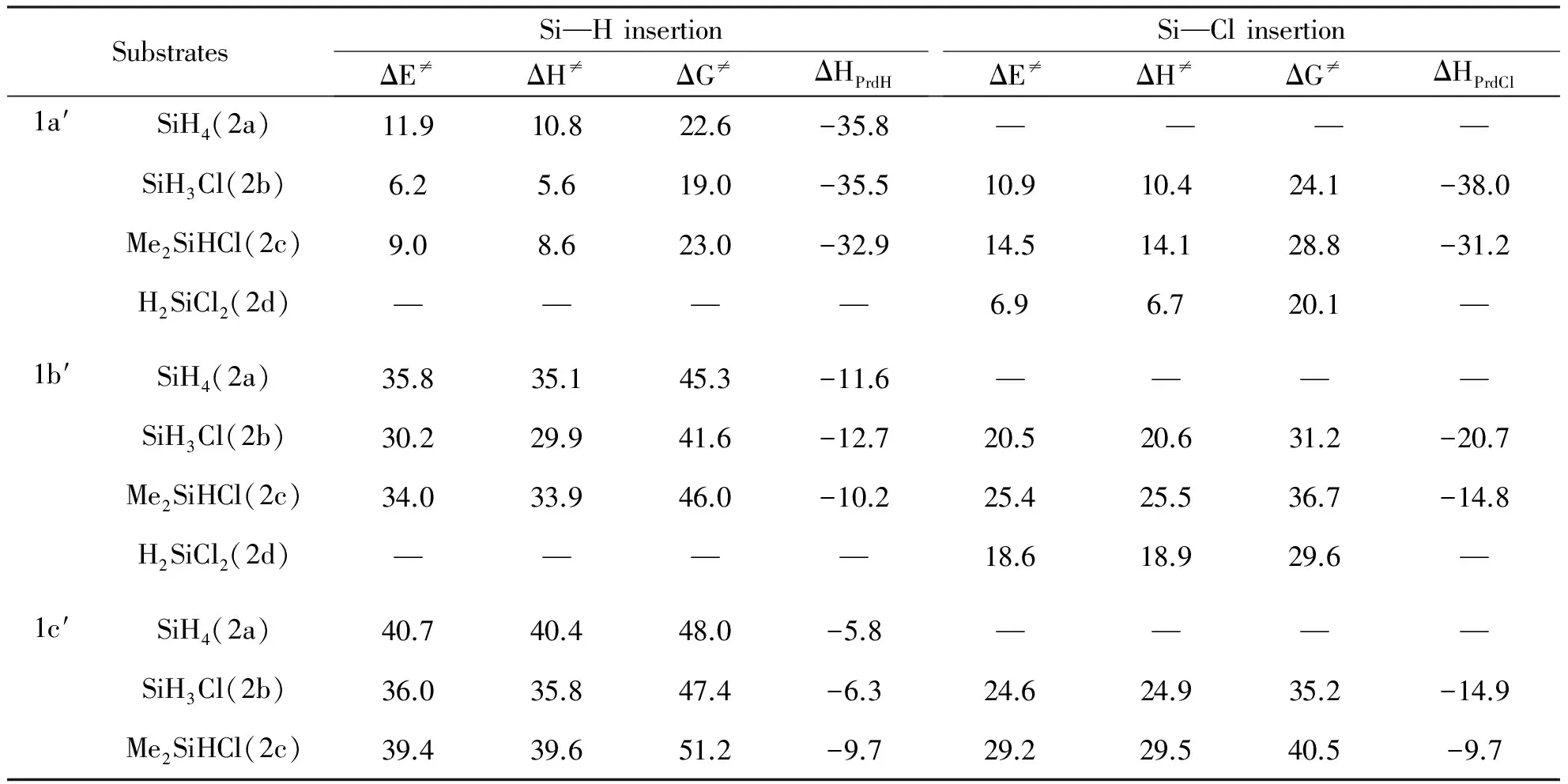
表1 B3LYP/6-31++G(d,p)水平上298.15 K下计算的硅烯1a′-1c′与氯硅烷2a-2c的Si—X(X=H,Cl)键插入反应中复合物、过渡态和产物的能量参数
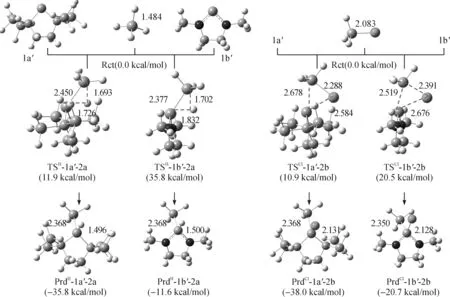
图2 B3LYP/6-31++G(d,p)水平上优化到的硅烯1a′和1b′分别与硅烷2a和2b发生Si—H和Si—Cl插入反应中的反应物、过渡态及产物的几何结构和能量参数Fig. 2 The optimized geometries of the stable points on PES for silylene 1a′ and 1b′ insertion into Si—H bond of SiH4 (2a) and Si—Cl bond of H3SiCl (2b) calculated at the B3LYP/6-31++G(d,p) level of theory

图3 B3LYP/6-31++G(d,p)水平上优化到的硅烯1a′和1b′分别与硅烷2c之间Si—H和Si—Cl插入反过渡态的几何结构和能量参数Fig. 3 The optimized TS geometries for silylene 1a′and 1b′ insertion into Si—H and Si—Cl bond of Me2SiHCl (2c)calculated at the B3LYP/6-31++G(d,p) level of theory
硅烯即有亲电性又有亲核性,在Si—X插入反应中硅烯与硅烷间的亲电和亲核轨道相互作用都对稳定过渡态的能量起贡献[10].我们推测二氨基取代硅烯对二甲基氢氯硅烷反应活性和选择性的改变与硅烯二价硅原子的亲电和亲核能力变化密切相关.硅烯1a′-1c′的二价硅原子的孤对电子轨道[n(Si)]和pπ空轨道[pπ(Si)]所在分子轨道的能量和形状示于图4中.从图中可以看出,二氨基取代硅烯中两个氮原子上的孤对电子会与pπ(Si)共轭,使pπ(Si)亲电性大为降低,表现为1b′和1c′的LUMO轨道较1a′分别上升了1.66 eV和1.26 eV.与pπ(Si)相比,n(Si)所在的分子轨道能量则只发生了小幅下降,1b′和1c′相应轨道分别下降了0.15 eV和0.61 eV.这说明二氨基取代硅烯的亲电性和亲核性都减弱了,且亲电性的减弱幅度显著大于亲核性.由此可见,二氨基取代硅烯键插入反应活化能的大幅度升高主要与它的亲电性减弱相关,Si—H插入反应活化能的增幅大于Si—Cl插入反应因硅烯的亲电作用对Si—H插入反应过渡态能量的稳定的贡献高达70%密切相关.

图4 硅烯1a′-1c′二价硅原子的孤对电子轨道n(Si)和pπ(Si)空轨道所在分子轨道的形状和能量(eV)Fig. 4 Molecular orbital shape of silylene ‘s(1a′-1c′) lone-pair orbital [n(Si)] and vacant pπ orbital [pπ(Si)]calculated at B3LYP/6-31++G(d,p) level of theory
此外,我们还比较了图5所示几组过渡态活化能垒,以此来了解硅烯的亲核作用对Si—X键插入反应活性的影响.从图5可以看到,将Si—H、Si—Cl键插入反应中与三元环过渡态处于同一平面的取代基由H更换为Cl时对应的活化能都发生了一定程度的降低(<5.7 kcal/mol).这种作用无法弥补由硅烯亲电性降低而给Si—H插入反应带来的降低反应活性的影响(16.7 kca/mol).
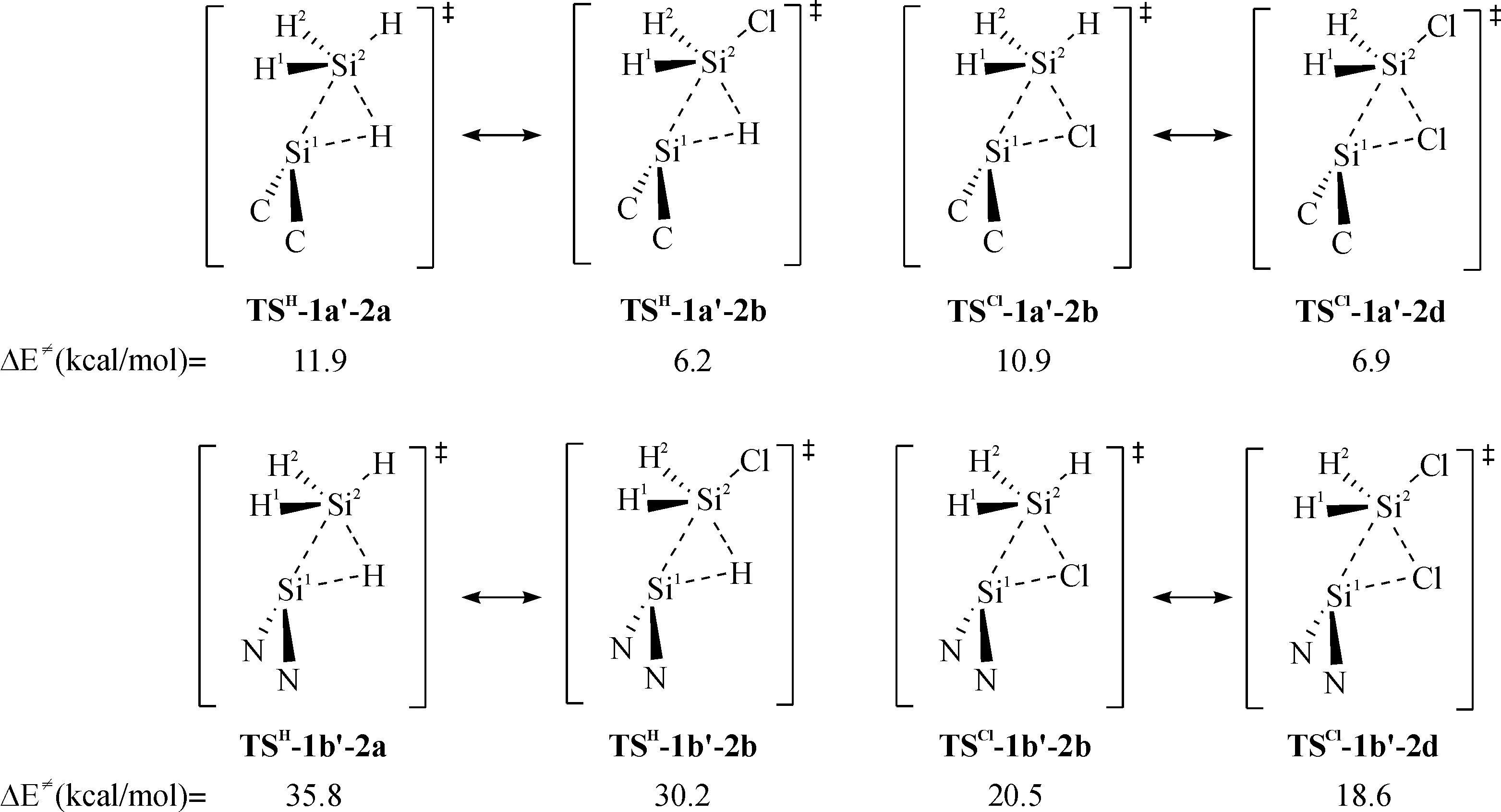
图5 硅烯1a′和1b′与硅烷2a、2b和2d发生Si—X(X=H或Cl)插入反应特定过渡态的活化能(kcal/mol)Fig. 5 Activation energy of selected TSs for the Si—X insertion reactions of silylene (1a′ and 1b′) with silane(2a, 2b, and 2d) (in kcal/mol)
3 结 论
上述3种硅烯与3种硅烷/氢氯硅烷的Si—X键插入反应势能面的计算和分析表明:(1) 二氨基取代硅烯的Si—X键插入反应的反应活性不高且放热不明显,产物在热力学上远不及二烷基取代硅烯的产物稳定;(2) 二氨基取代硅烯对二甲基氢氯硅烷(Me2SiHCl)表现出了与二烷基取代硅烯不同的反应选择性,Si—Cl插入反应优先于Si—H插入反应;(3) Si—H插入反应对硅烯亲电性的依赖性大,亲电性降低会使该反应的活化能大幅度增高,使得由硅烷中Si原子上的吸电子取代基与硅烯间的增强的亲核作用无法像亲电性强的硅烯那样对反应选择性产生影响.亲电性差的硅烯与各种氢氯硅烷反应时,Si—Cl键插入反应优先发生.如要达到使硅烯与氢氯硅烷的Si—H键插入反应优先发生的目的,需从提高硅烯的亲电性着眼.
[1]Gasper P P, Herold B J.Silicon, germanium and tin structural analogs of carbenes? [M]// Kirmse W. Carbene Chemistry. New York: Academic Press,1971:504-550.
[2]Tokitoh N, Ando W. Silylenes (and germylenes, stannylenes, plumbylenes)[M]// Moss R A, Platz M S, Jr Jones M .Reactive Intermediate Chemistry. Hoboken, New Jersey: John Wiley & Sons, Inc,2004:651-716.
[3]Chen Chihui, Su Mingder. The mechanism of C-X (X = F, Cl, Br, and I) bond activation in CX4by a stabilized dialkylsilylene [J]. Chem Eur J,2007,13(24):6932-6941.
[4]Gehrhus B, Hitchcock P B, Jansen H. The stable silylene Si (NCH2But)(2)C6H4-1,2 : reactions with group 14 element halides [J]. J Organomet Chem,2006,691(4):811-816.
[5]Herzog U, Richter R, Brendler E,etal. Methylchlorooligosilanes as products of the basecatalysed disproportionation of various methylchlorodisilanes[J]. J Organomet Chem,1996,507:221-228.
[6]Gaspar P P. Reactive intermediates Vol. 3 [M]. Jr Jones M, Moss R A, Eds. New York :Wiley,1985:333-427.
[7]Auner N, Weis J. Organosilicon Chemistry Ⅳ[M]. VCH: Weinheim, Germany,2000.
[8]Michael H, Thomas A S, Robert W. Stable silylenes[J]. Acc Chem Res,2000,33(10):704-714.
[9]Ishida S, Iwamoto T, Kabuto C,etal.Insertion of a stable dialkylsilylene into silicon-chlorine bonds [J]. Silicon Chem,2003,2(3/4):137-140.
[10]Xu Zheng, Jin Juan, Li Zhifang,etal. Remarkable substituent effects on the activation energy of silylene insertion into silicon-chlorine bonds [J]. Chem Eur J,2009,15(34):8605-8612.
[11]Kohn W, Becke A D, Parr R G. Density functional theory of electronic structure[J]. J Phys Chem,1996,100(31):12974-12980.
[12]Gonzalez C, Schlegel H B. An improved algorithm for reaction-path following[J]. J Chem Phys,1989,90(4):2154-2161.
[13]Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03, revision D. 01 [CP]. Gaussian, Inc.: Wallingford,2004.
SelectiveInsertionReactionofSilyleneintoSi—XBondofHydrochlorosilane
YANG Xuemin, JIN Juan, XU Zheng, LI Zhifang, QIU Huayu, JIANG Jianxiong, LAI Guoqiao
(1Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University,Hangzhou 310012, China)
Calculating the potential energy surface of the insertion reaction of σ bond among three silylenes and four silane/chlorosilane modle moleculars, the paper studied the reaction selectivity for steric stabilzed dialkylsilylene and electronic stabilized diaminosilylene insertion into Si—X (X=H, Cl) bond of hydrochlorosilanes by B3LYP/6-31++G(d,p) method. The results indicate that the reactivity of diaminosilylene is apparently lower, and the reaction is less exothermic. The Si—Cl insertion is preferred to the Si—H insertion, the reason of which is that the stabilization of transition state energy in Si—H insertion reaction has more dependence on the electrophilicity of silylene than Si—C1 insertion reaction. The stronger dependence of Si—H insertion barrier to silylene’s electrophilicity is considered as the main reason.
theoretical calculation; silylene; hydrochlorosilane; Si—X insertion reaction
2012-11-01
国家青年科学基金项目(20903032);浙江省自然科学基金项目(Y4090470).
徐 征(1978—),女,副研究员,主要从事有机硅化学相关反应机理的理论和实验研究.E-mail:zhengxu@hznu.edu.cn
10.3969/j.issn.1674-232X.2013.01.003
O64
A
1674-232X(2013)01-0014-07
