基于PCR-DGGE的小鼠肠道菌群基因组提取方法的建立
2013-10-25张素珍李思施韦婷婷关家伟吴大畅
张素珍,李思施,韦婷婷,张 凯,关家伟,吴大畅
(大连医科大学生物技术系,辽宁 大连 116044)
哺乳动物肠道内细菌组成非常复杂。近年来,肠道菌群对机体健康和疾病的影响越来越受到人们的关注[1-2],粪便与肠道内容物相似,含有大量的微生物、未消化食物和肠道脱落细胞等[3],粪便微生物的变化可以间接地反映肠道微生物的变化。粪便分析技术不同于采血或取组织获取样本,属于非损伤性取样,应用性强[4]。在肠道菌群数量和种类鉴定过程中,因厌氧菌占多数,难以利用传统的选择培养法进行培养,因此无法对其深入研究[5]。近年来,分子微生态学技术被广泛应用于肠道菌群多样性研究中,变性梯度凝胶电泳(DGGE)通过递增的化学变性剂浓度梯度把长度相同碱基组成不同的DNA片段区分开。该法可有效避免在传统富集、培养、分离研究过程中造成的微生物多样性丢失,能够更直接、可靠地反映出微生物原始组成情况[6]。然而能否获得高浓度、多样性程度高、具有代表性的微生物基因组DNA对后续的PCR及DGGE分析具有至关重要的作用。粪便中大量食物残渣、组织脱落物和各种消化道成分的存在,对微生物DNA的提取造成很大困难。目前粪便微生物细胞的裂解方法主要有化学裂解法、物理裂解法、酶解法[7]。不同方法对微生物细胞的裂解程度不尽相同,可根据实验目的的不同将以上方法有机结合以达到最佳结果。本研究在总结以往报道的基础上,对4种小鼠粪便微生物总DNA提取方法进行比较和研究,进一步加以改进,摸索出适用于小鼠粪便细菌基因组DNA提取的有效方法,旨在为后续基于PCR扩增及DGGE分析肠道菌群结构提供前提基础和实验保障。
1 材料与方法
1.1 材料
1.1.1 样品采集 SPF、BALB/C小鼠由大连医科大学实验动物中心提供,3周龄,体重18~22 g。肛门直接收集粪便样品于灭菌Ep管中,备用。
1.1.2 主要试剂及仪器 PCR引物由大连宝生物公司合成,Ex Taq DNA聚合酶购自大连宝生物公司,粪便DNA提取试剂盒(市售,中国)。丙烯酰胺、甲叉双丙烯酰胺购自美国Sigma公司,去离子甲酰胺购自上海生物化学试剂工程公司。DCode变性梯度凝胶电泳系统(大连竞迈生物科技有限公司),TDL5M台式低速大容量冷冻离心机(长沙湘智离心机仪器有限公司)、空气恒温摇床 KYC-100B、Thermo PCR 循环仪(PXE 0.2 Thermal Cyder)、PHs-3C精密pH计(上海精密科学仪器有限公司),超微量分光光度计(NanoVue,USA)。
1.2 方法
1.2.1 粪便样品总DNA的提取 ①基于SDS的裂解法[8-9](方法Ⅰ):取粪便样品0.2 g于盛有2 mL PBS的离心管中,混匀,3000 r/min离心10 min,重复1次。加入1.5 mL的DNA提取液(100 mmol/L Na2EDTA,100 mmol/L 磷酸钠,100 mmol/L Tris-HCl,1.5 mol/L NaCl,1%CTAB,pH 8.0)和5 μL蛋白酶K(40 mg/mL)于37℃摇床中140 r/min,1 h;加入115 μL 20%SDS,65 ℃水浴2 h(每隔15~20 min轻摇1次),于室温4000 r/min离心10 min,取上清。再向沉淀中加入720 μL DNA提取液、2 μL 蛋白酶 K 及 80 μL 20%SDS,漩涡震荡10 s,65℃水浴10 min,4000 r/min离心10 min,取上清。混合2次上清液;②市售国产试剂盒提取DNA(方法Ⅱ):严格按照试剂盒操作说明规范操作;③改进的化学裂解法 (方法Ⅲ):称取0.2 g小鼠粪便,加入1 mL PBS,漩涡震荡,充分混匀后,200×g离心5 min,取上清。重复1次,混合上述2次上清,300×g离心5 min。将上清转移到新的Ep管中,10000 r/min离心8 min,沉淀菌体。将所得的菌体用1 mL PBS洗涤2~3次(至上清无色),-20℃保存备用。向Ep管中加500 μL DNA 提取液(同①),6 μL 蛋白酶 K(40 mg/mL)37℃、140 r/min摇床振荡1 h左右,再加入56 μL 20%SDS,65℃水浴2 h(每隔15~20 min轻摇1次);④改进的溶菌酶法[10](方法Ⅳ):同③的方法沉淀菌体。参考改进溶菌酶法[10],稍作改动。方法Ⅰ和Ⅲ得到的细胞裂解液均用等体积的酚/氯仿(Tris-饱和酚∶氯仿∶异戊醇=25∶24∶1)进行抽提2~3次,以除蛋白,以0.6倍体积的异丙醇于室温下沉淀DNA,12000r/min离心10 min,75%乙醇洗涤沉淀,DNA沉淀自然干燥后溶于TE(加RNAse)溶液中并于37℃温箱中放置30 min。1%琼脂糖凝胶电泳检测。
1.2.2 16S rDNA V3区的 PCR扩增 以上述DNA提取液为模板扩增所有细菌16S rDNA基因的V3可变区。根据文献[11]设计引物及扩增体系。PCR反应程序:预变性94℃ 5 min;变性94℃ 30 s,退火54℃ 30 s,延伸72 ℃ 30 s,30个循环;72℃ 延伸7 min。扩增结果1%琼脂糖凝胶电泳检测。
1.2.3 PCR反应产物的变性梯度凝胶电泳(DGGE)分析 35% ~55%的变性范围,60℃恒温水浴,电压50~60 V,电泳4 h。电泳完毕后,EB染色30 min,观察并拍照。利用Gel-Pro analyzer(灰度分析)软件分析DGGE图谱。获得各泳道内各条带的IOD,然后利用Shannon-Weaver index(H’)计算条带的多样性[12]。并采用均匀度Evenness(E)分析菌群分布的统一性。H’和E通过以下公式获得:
Shannon-Weaver index(H’)=-∑(Pi)(ln Pi);Evenness(E)=H’/InS Pi=ni/N,其中ni为单个条带的灰度值,N为所有条带的灰度值,S为样品条带数。
2 结果与分析
2.1 4种方法提取DNA质量的比较
2.1.1 琼脂糖凝胶电泳检测结果 4种方法对同一粪便进行DNA提取的琼脂糖凝胶电泳检测结果见图1。方法Ⅰ和方法Ⅱ所得DNA提取液在20 kb左右均未见清晰条带,提示未能成功提取细菌基因组总DNA。而方法Ⅲ和Ⅳ在目标位置可见清晰条带。图中泳道1~3、7、8主条带亮度虽较高,但有少许弥散,说明DNA提取率高但纯度不够,泳道4~6条带亮度虽不高,但泳道清晰,无弥散。2种方法均成功地提取了细菌基因组。
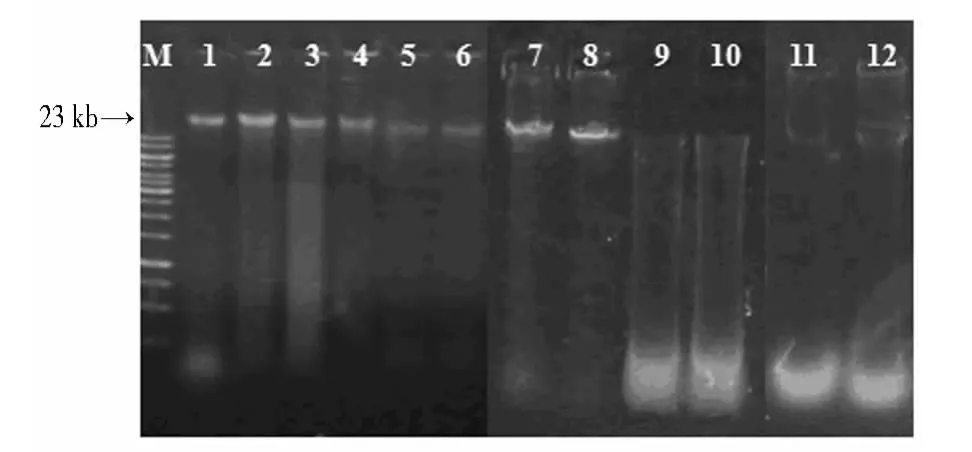
图1 粪便细菌总DNA提取的琼脂糖凝胶电泳检测Fig.1 Agarose gel electrophoresis detection of total DNA from fecal bacteria
2.1.2 方法Ⅲ和Ⅳ 两种方法所得DNA的纯度和浓度比较 OD260nm/OD280nm能够反映DNA的纯度,正常 OD260nm/OD280nm值约为 1.8 ~2.0。若OD260nm/OD280nm值小于1.8或大于2.0说明可能有蛋白质或RNA污染[10],2种DNA提取法获得DNA的纯度和浓度见表1。

表1 DNA纯度和浓度检测结果Table 1 Detection of DNA purity and concentration
2.2 细菌DNA的16S rDNA V3区 PCR扩增结果
由于方法Ⅰ和方法Ⅱ未能成功提取到肠道菌群总DNA,故不能进行后续的PCR扩增,所以只对方法Ⅲ和方法Ⅳ所提取的细菌总DNA进行PCR扩增。由图2可知,2种方法PCR产物的大小约为230 bp,说明由2种方法提取的DNA均可得到较理想的PCR结果。
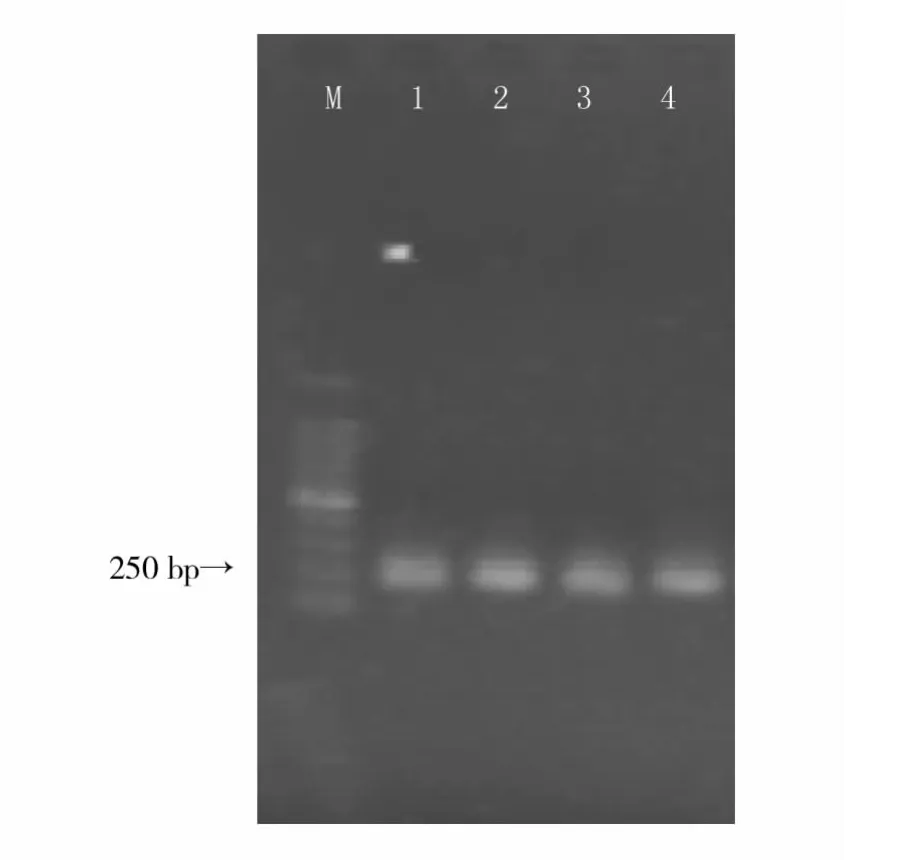
图2 PCR产物的琼脂糖凝胶电泳结果Fig.2 Result of PCR amplication
2.3 菌群多样性DGGE结果
在从粪便提取细菌总DNA的过程中不可避免地会使一些数量较少的劣势菌丢失,造成菌群多样性降低。DGGE图谱能够直观地反映出菌群多样性,进而判断DNA提取方法的优劣。由图3分析得出,2种方法均能分离出粪便20余种细菌。
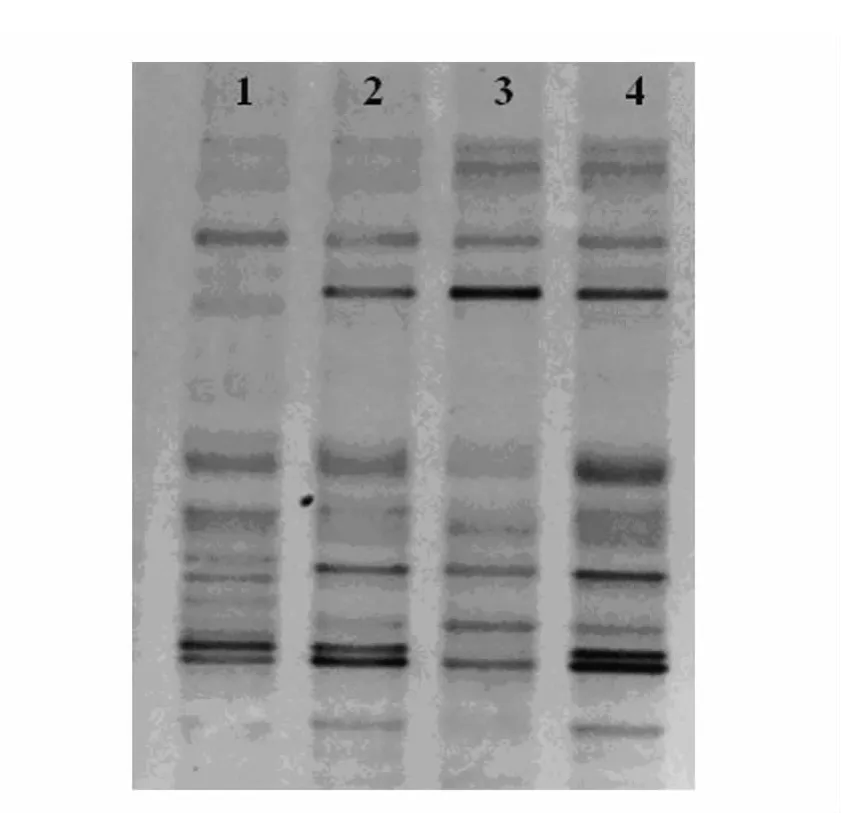
图3 DGGE结果Fig.3 Result of DGGE
利用Gel-Pro analyzer(灰度分析)软件分析DGGE胶图及统计学分析数据所得的丰富度(S)、多样性(H)及均匀性(E)的结果见表2。由表2可知,方法Ⅲ和方法Ⅳ的多样性和均匀性均较好,但方法Ⅳ的丰富度更好。另外利用方法Ⅲ提取DNA的相同样品之间的差异性较大,较方法Ⅳ的稳定性差。

表2 DGGE图谱多样性分析结果Table 2 Diversity analysis of DGGE fingerprinting
3 讨论
粪便中含有较多杂质,在粪便微生物DNA提取过程中,未消化的食物残渣、黏液、消化酶等杂质可能会导致DNA降解或后续PCR反应抑制物的产生。因此,在动物粪便微生物总DNA提取时,保证DNA的纯度和浓度是提取技术的关键。目前,各种提取粪便细菌总DNA的方法均是围绕如何去除这些杂质和抑制物进而得到高纯度且足够量的目的基因而展开[13]。在化学裂解法裂解微生物细胞过程中,通常包括能够攻击细菌细胞壁和除去细胞膜的2种成分,从而暴露DNA。溶菌酶和乙二胺四乙酸盐(EDTA)通常用于削弱细胞壁的作用,去垢剂如十二烷基硫酸钠(SDS)来破坏细胞膜,协助整个细胞外壁的裂解过程[14]。另外阳离子表面活性剂十六烷基三甲基溴化铵(CTAB)主要功能是分解多糖,破坏细胞壁完整性。在DNA提取中常用的蛋白酶K能够降解蛋白,使DNA充分游离。
本实验采用基于SDS的裂解法(Ⅰ)、国产市售试剂盒(Ⅱ)、改进的化学裂解法(Ⅲ)和改进的溶菌酶法(Ⅳ)四种方法分别对小鼠粪便细菌总DNA进行了提取,其中方法Ⅲ是在方法Ⅰ的基础上改进而来。方法Ⅰ和方法Ⅱ是直接在粪便中加入细胞裂解液,而方法Ⅲ和方法Ⅳ则是先将微生物从粪便杂质中分离除杂后再裂解微生物细胞。从DNA提取结果可以看到,先分离后裂解的效果优于直接裂解细胞。本研究还发现,分离除杂后的微生物细胞经过反复冻融后再进行裂解的DNA提取效果较分离除杂后直接裂解效果好。在方法Ⅰ和方法Ⅲ中:通过EDTA、CTAB及蛋白酶K的配合使用,达到破碎微生物细胞,获取游离DNA的目的。方法Ⅳ则利用溶菌酶破坏细胞壁。从DNA提取的浓度和纯度上看,方法Ⅲ提取的DNA纯度高于方法Ⅳ,但DNA的浓度却低于方法Ⅳ。OD260nm/OD280nm显示方法Ⅲ所得DNA提取液中含有一定量的RNA杂质,而方法Ⅳ则含有蛋白质污染。但2种方法得到的总DNA均能成功地进行后续PCR扩增,且变性梯度凝胶电泳图谱中清晰地分出了20多种不同的条带。除多样性和均匀性相似外,方法Ⅳ的丰富度和稳定性更佳。
目前多数实验室采用市售试剂盒提取粪便菌DNA,效果虽好但价格昂贵,而常规的传统DNA提取方法步骤多、耗时长,且提取质量不佳,二者均不适合基层实验室大批量分析使用,故如何利用分子生物学常规试剂建立粪便细菌总DNA提取的稳定、经济、快捷的方法,并能够适用于下游PCR-DGGE分析具有重要的实践和经济意义。本研究中的方法Ⅲ和Ⅳ利用实验室常规试剂完成了对肠道菌群总DNA的较高质量的提取,成本低且重复性好,具有较强的实用价值,为研究肠道菌群的结构提供了基础,值得实验室深入推广。
[1]Nicholson J K,Holmes E,Wilson I D.Gut microorganisms,mammalian metabolism and personalized health care[J].Nature Reviews Microbiology,2005,3(5):431-438.
[2]李凯,谭永刚,李午生,等.双歧杆菌预防化疗后肠道菌群失调症的临床研究[J].微生物学杂志,2011,31(1):82-84
[3]魏辅文,饶刚,李明,等.分子粪便学及其应用—可靠性、局限性和展望[J].兽类学报,2001,21(2):143-151.
[4]徐敏娟,王志跃.微生物总 DNA的提取[J].草业科学,2008,25(5):78-80.
[5]卢龙娣,江胜滔,林跃鑫.鸡肠道菌群总 DNA三种提取方法的比较[J].中国微生态学杂志,2009,21(7):588-590.
[6]Muyer G,Waal E C de,Ultterlinden A G.Profiling of complex microbial population by denaturing gradient gel electrophoreses analysis of polymerase chain reaction-amplified genes coding for 16S rRNA[J].Appl Environment Microbiology,1993,59(3):695-700.
[7]张珍妮,吴晓芙,陈永华,等.DGGE技术在环境微生物多样性研究中的应用[J].生物技术通报,2009,7(12):48-51.
[8]李鹏,毕学军,汝少国.DNA提取方法对活性污泥微生物多样性PCR-DGGE检测的影响[J].安全与环境学报,2007,2:53-54.
[9]吴银宝,史金才,莫测辉,等.猪粪和土壤样品中微生物DNA提取方法的比较[J].农业工程学报,2006,22(增刊2):10-13.
[10]郑刚,陈己任,赵国华,等.基于DGGE分析的大鼠粪便及肠道细菌DNA提取方法研究[J].生物工程,2011,32(17):215-216.
[11]卢珊,孙晖,熊衍文,等.婴幼儿腹泻粪便标本菌群的16S rDNA PCR-DGGE 分析[J].中国人兽共患病学报,2009,25(3):225-228.
[12]Silva EP,Russo CAM.Techniques and statistical data analysis in molecular population genetics[J].Hydrobiologia,2000,420:119-135.
[13]罗勇军,刘听.实时荧光定量PCR标准品的制备及应用[J].重庆医学,2005,34(3):414.
[14]T.A.Brown.Gene cloning and DNA analysis[M].2003.
