CCl4诱导的小鼠纤维化肝组织微小RNA差异表达谱及初步功能分析*
2013-10-24王清兰李俊霞陶艳艳闫秀川刘成海
王清兰, 李俊霞, 吕 靖, 陶艳艳, 闫秀川, 刘成海, 2, 3, 4△
(1上海中医药大学附属曙光医院肝病研究所, 2上海市中医临床重点实验室, 3上海中医药大学肝肾疾病病证教育部重点实验室, 4上海高校中医内科学E研究院,上海 201203)
CCl4诱导的小鼠纤维化肝组织微小RNA差异表达谱及初步功能分析*
王清兰1,2, 李俊霞1, 吕 靖1, 陶艳艳1, 闫秀川1, 刘成海1, 2, 3, 4△
(1上海中医药大学附属曙光医院肝病研究所,2上海市中医临床重点实验室,3上海中医药大学肝肾疾病病证教育部重点实验室,4上海高校中医内科学E研究院,上海 201203)
目的应用微小RNA(miRNA)芯片技术研究miRNAs在四氯化碳(CCl4)诱导的小鼠纤维化肝脏中的差异表达谱,并基于基因本体论(gene ontology,GO)分析及信号转导通路分析发现差异miRNAs的主要功能。方法实验分为正常组及模型组,皮下注射 CCl4复制小鼠肝纤维化模型;应用Agilent 小鼠 miRNA 寡核苷酸基因芯片检测各组肝脏miRNA表达谱。用随机方差模型t检验筛选2组间的差异miRNAs,并预测其靶基因。对靶基因进行GO分析及信号转导通路分析发现差异miRNAs发挥的主要功能。结果正常组与模型组间共筛选出39个差异miRNAs,其中模型组较正常组上调的23个,下调的16个。GO分析及信号转导通路分析结果提示差异miRNAs可能调控的靶基因及其参与的生物学功能包括细胞的增殖与活化、细胞凋亡、细胞周期、细胞黏附、细胞迁移、炎症反应、转化生长因子β(TGF-β)/Smads信号转导通路、Wnt受体信号转导通路、蛋白代谢过程的调控等。GO分析发现关键的上调miRNA包括mmu-miR-322、mmu-miR-15b、mmu-miR-195、mmu-miR-200b、mmu-miR-214等,关键的下调miRNA包括mmu-miR-16、mmu-miR-130a、mmu-miR-101b、mmu-miR-30a和mmu-miR-30e等。对显著性GO与显著性信号通路所属的靶基因取交集,对网络中miRNA在网络中的调控地位进行评价,结果发现关键的上调miRNAs包括mmu-miR-200b、mmu-miR-322、mmu-miR-106b、mmu-miR-23a、mmu-miR-15b等,关键的下调miRNAs包括mmu-miR-16、mmu-miR-30e、mmu-miR-30c、mmu-miR-30a、mmu-miR-130a等。结论纤维化肝组织miRNAs表达较正常肝组织发生明显变化;肝纤维化形成的各个环节,包括细胞的增殖与活化、细胞黏附、细胞凋亡、细胞迁移与分化、物质代谢、TGF-β信号通路等都可能受miRNAs的调控。
微小RNA; 肝纤维化; 差异表达; 四氯化碳
微小RNA(microRNA,miRNA)是一类内源性非蛋白质编码的RNA分子,长约20~25个核苷酸[1], 广泛存在于各种动植物甚至单细胞真核生物中。miRNA通过直接剪切靶mRNA,或者通过完全/不完全与靶mRNA 3’非翻译区互补结合抑制靶mRNA的翻译。miRNA可以与不完全互补的mRNA配对来抑制蛋白质的翻译,每个miRNA可以有多个靶基因,而同一个基因也可以同时受几个miRNAs的调节。这种复杂的调节网络既可以通过一个miRNA来经济地调控多个基因的表达,也可以通过几个miRNAs的组合来精细调控某个基因的表达。越来越多的实验结果表明miRNA有着调控细胞生理学几乎所有方面的功能潜力,包括胚胎发育、细胞增殖及死亡、细胞凋亡与分化、脂肪代谢等[2-6],而且具有特异的时空表达特点,参与了人类多种生理病理改变的过程。大量研究表明[7],miRNA在部分脏器慢性损伤,特别是肝脏的慢性损伤过程中存在差异。因此,本研究采用miRNA芯片,研究miRNA在四氯化碳(carbon tetrachloride,CCl4)诱导的小鼠纤维化肝脏中的表达特点。
材 料 和 方 法
1材料
1.1动物 雄性C57BL/6小鼠,23~25 g,SPF级,购于中国科学院上海实验动物中心,许可证号为SYXK(沪)2007-0005。所有小鼠饲养于上海中医药大学动物实验中心,自由饮食。
1.2主要试剂 CCl4(分析纯)和橄榄油(分析纯)购自国药集团化学试剂有限公司;总RNA抽提试剂盒(mirVanaTMRNA Isolation Kit)为Life Technologies产品; 样品标记及杂交试剂盒(miRNA Complete Labeling and Hyb Kit)和小鼠miRNA 寡核苷酸基因芯片试剂盒(Mouse miRNA Microarray Kit 12.0)均为Agilent产品。
1.3主要仪器 芯片扫描仪(Microarray Scanner, Agilent G256BA), 杂交盒(Hybridization Chamber, Agilent G2534A),杂交炉(Hybridization Oven, Agilent G2545A), SpectraMax M5 酶标仪。
2方法
2.1分组及模型制备 23只小鼠随机分为正常组(n=10)及模型组(n=13)。模型制备按参考文献方法并加以改进[8],首次以CCl43 mL/kg体重皮下注射,以后以50% CCl4-橄榄油3 mL/kg体重皮下注射,1周2次,共8周。
2.2样品的采集与处理 小鼠经摘眼球采血,3 000 r/min离心15 min,收集血清,-70 ℃保存;断颈处死后,打开腹腔,摘取肝脏;取0.8 cm×0.8 cm×0.3 cm大小肝组织2块,1块以10%甲醛液固定,24 h后逐级乙醇脱水,二甲苯透明,60 ℃石蜡包埋,用于普通病理及免疫组化实验;其余组织切碎后入液氮速冻,-70 ℃保存,用于miRNA芯片检测。
2.3肝组织病理染色 肝组织经4%甲醛液固定,石蜡包埋,4 μm切片,二甲苯、多级乙醇脱蜡至水,做天狼星红染色。肝组织天狼星红染色后参照文献进行肝纤维化分级[9]。
2.4肝组织羟脯氨酸(hydroxyproline, Hyp)含量测定 参照Jamall等[10]盐酸水解法。100 mg湿肝组织,加12 mol/L HCl在105 ℃水解18 h。过滤,取100 μL水解液40 ℃烘干。同时取Hyp标准品0.2~1.6 μg设为标准对照管,经氯胺T溶液0.2 mL,欧式液[含25%(W/V)对二甲基氨基苯甲醛和27.3%(V/V)高氯酸的异丙醇溶液]反应,50 ℃水浴90 min,读取A558值,计算标准曲线。同上方法测定肝组织样本A558值,根据标准曲线计算样本含量,用肝组织湿重校正。
2.5组织总RNA (含miRNA)提取 称取约50 mg肝组织,按照mirVanaTMRNA Isolation Kit试剂盒说明提取总RNA。采用Agilent 2100 Bioanalyzer核酸质检系统检测RNA样本的浓度及质量。质控标准为:RIN(RNA integrity number)≥6.0且28S/18S>0.7。
2.6miRNA芯片杂交及扫描 miRNA芯片杂交及扫描均由生物芯片上海国家工程研究中心完成。取RNA 100 ng,加入去磷酸化混合液,37 ℃孵育30 min进行去磷酸化处理。随后向样品中加入DMSO,100 ℃金属浴5~10 min后,迅速放入冰水浴中。向冰浴后的样品中加入连接反应混合液,16 ℃温育2 h。反应结束后,将样品置于真空浓缩仪中完全抽干,重新溶解在无核酸酶去离子水中,加入10×GE 封闭溶液和2×Hi_RPM 杂交缓冲液,100 ℃金属浴5 min后冰浴5 min。准备杂交反应混合液,组装杂交舱。将杂交舱平衡放置在杂交炉的架子上,55 ℃、20 r/min杂交20 h。杂交结束后,洗涤芯片。芯片采用Agilent扫描仪进行扫描,Agilent Feature Extraction 软件读取数据,扫描分辨率5 μm, PMT 100%,5%,采用Feature Extraction 进行处理分析。最后应用GeneSpring 10.0 进行quantile normalization。
2.7芯片数据分析 (1)采用随机方差模型[11-12]筛选正常组与模型组的差异miRNAs (TwoClassDif)。(2)通过搜索mirdb数据库,得到差异miRNAs调控的所有靶基因。(3)对差异miRNAs对应的靶基因进行基因本体论(gene ontology,GO)分析[13-14],得到具有显著性、低误判率、靶向性的功能以及对应的靶基因。采用下列公式进行GO的富集度分析。
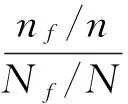
Nf:差异基因的数目;
n:GO中含有基因的总数目;
N:芯片上检测出来基因的总数目。
(4)对差异miRNAs的靶基因进行Pathway显著性分析[15-16],得到具有显著性、低误判率、靶向性的Pathway以及对应的靶基因。Pathway富集度分析同GO的富集度分析。(5)对靶基因的显著性功能和显著性Pathway分析,将显著性GO和Pathway所包含的靶基因取交集。(6)构建差异miRNAs与交集靶基因的调控网络(miRNA-gene network),得到网络中起核心调控作用的miRNA和被miRNA调控的关键靶基因[17-18]。
用差异miRNA调控的靶基因与miRNA的属性关系来建立miRNA-基因作用网络。通过基因与miRNA的属性关系可以建造基因与miRNA的邻接矩阵A=[aij],aij表示基因i与miRNA j的关系权重。在miRNA与基因的网络中,基因用圆圈来表示,miRNA用方形来表示,相互作用用直线来表示。网络中心用网络的度来表示。度表示miRNA对周围基因的贡献程度,或基因对周围miRNA的贡献程度。核心miRNA或基因就是在网络图中度最高的。
3统计学处理
计量资料用均数±标准差(mean±SD)表示。使用SPSS 12.0软件包分析,组间比较使用单因素方差分析。纤维化分级计数资料采用Ridit分析, miRNA差异基因筛选采用随机方差模型的t检验,以P<0.05为差异有统计学意义。GO和Pathway分析采用Fisher精确检验和2检验。
结 果
1模型小鼠血清肝功能指标和肝组织Hyp含量变化
与正常组相比,模型组小鼠血清丙氨酸氨基转移酶(alanine aminotransferase,ALT)和天冬氨酸转氨酶(aspartate aminotransferase,AST)明显升高 (P<0.01),肝组织Hyp含量明显升高 (P<0.01),见图1。

Figure 1. Changes of liver function and Hyp between normal group and model group.Mean±SD.**P<0.01vsnormal group.
图1模型小鼠血清肝功能指标和Hyp含量的变化
2模型小鼠肝组织炎症病理及胶原沉积变化
HE染色结果显示:正常组肝细胞以中央静脉为中心呈放射状排列,形成肝板结构,细胞形态完整,未见坏死,胞核居中,无明显炎症细胞浸润;模型组肝脏汇管区大量肝细胞溶解性坏死,核溶解,网状支架塌陷,小叶结构破坏,并可见网状纤维周围大量肝细胞气球样变性、脂肪样变及炎症细胞浸润,见图2。
天狼星红染色结果显示,正常组汇管区可见少量胶原沉积;与正常组相比,模型组正常小叶结构破坏,汇管区胶原纤维沉积、延伸,相互交联形成多个假小叶结构,见图2。Ridit分析结果显示,与正常组相比,模型组小鼠肝组织纤维化分级Ridit值明显升高(P<0.01),见表1。
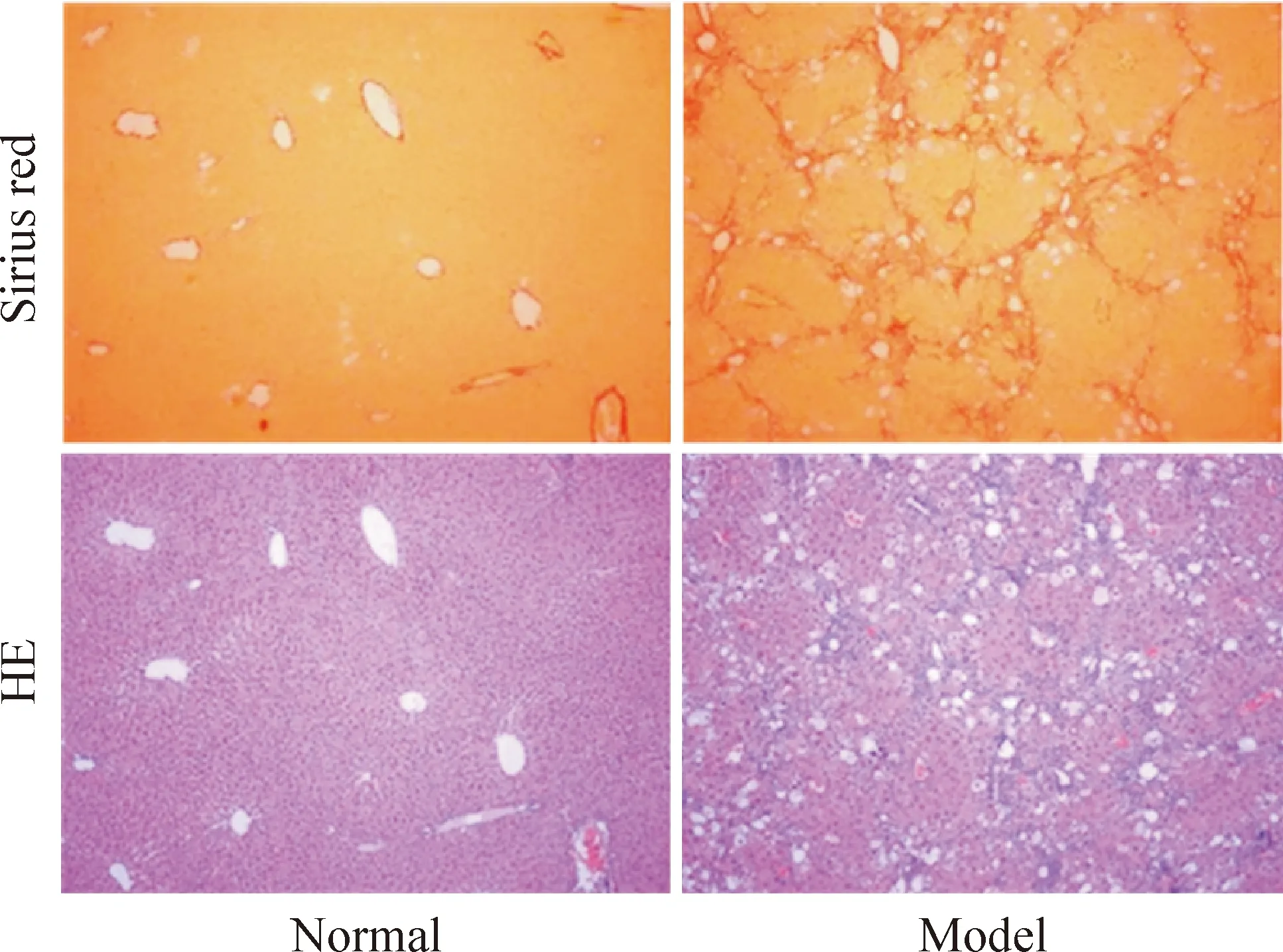
Figure 2. Changes of inflammation (HE staining) and collagen deposition (Sirius red staining) in liver tissues (×100).
图2模型小鼠肝组织炎症及胶原沉积的变化

表1 各组小鼠肝组织胶原纤维程度半定量分级
**P<0.01vsnormal group.
3模型组较正常组差异表达的miRNAs筛选
用随机方差模型对正常组与模型组的差异miRNAs(TwoClassDif)进行分析,P<0.05被认为有显著差异,共筛选到39个差异miRNAs,其中模型组较正常组上调的23个(表2),下调的16个(表3)。
4差异miRNAs靶基因的GO分析
模型组较正常组上调的miRNAs所对应的靶基因共参与80项显著性功能(图3),包括蛋白氨基酸磷酸化作用、对细胞增殖的负向/正向调控、细胞黏附、对细胞移行的正向调控、对凋亡的负向调控、细胞周期、肝脏发育等;模型组较正常组下调的miRNAs所对应的靶基因共参与54项显著性功能(图4),包括染色质改变、低氧应答、蛋白氨基酸磷酸化、对细胞增殖的负向/正向调控、细胞黏附等。
表2模型组较正常组表达上调的miRNAs
Table 2. The up-regulated miRNAs in model group compared with normal group
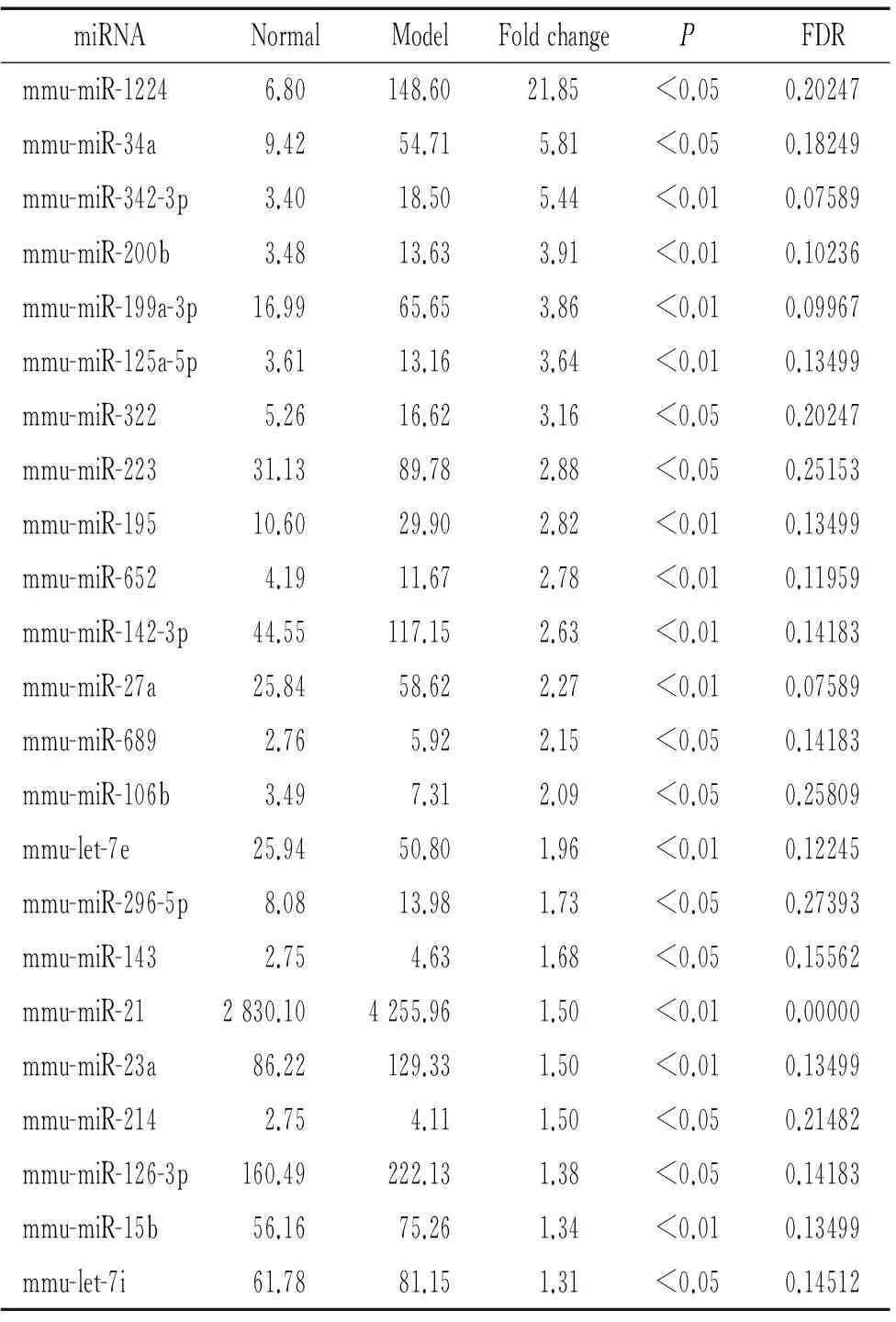
miRNANormalModelFoldchangePFDRmmu-miR-12246.80148.6021.85<0.050.20247mmu-miR-34a9.4254.715.81<0.050.18249mmu-miR-342-3p3.4018.505.44<0.010.07589mmu-miR-200b3.4813.633.91<0.010.10236mmu-miR-199a-3p16.9965.653.86<0.010.09967mmu-miR-125a-5p3.6113.163.64<0.010.13499mmu-miR-3225.2616.623.16<0.050.20247mmu-miR-22331.1389.782.88<0.050.25153mmu-miR-19510.6029.902.82<0.010.13499mmu-miR-6524.1911.672.78<0.010.11959mmu-miR-142-3p44.55117.152.63<0.010.14183mmu-miR-27a25.8458.622.27<0.010.07589mmu-miR-6892.765.922.15<0.050.14183mmu-miR-106b3.497.312.09<0.050.25809mmu-let-7e25.9450.801.96<0.010.12245mmu-miR-296-5p8.0813.981.73<0.050.27393mmu-miR-1432.754.631.68<0.050.15562mmu-miR-212830.104255.961.50<0.010.00000mmu-miR-23a86.22129.331.50<0.010.13499mmu-miR-2142.754.111.50<0.050.21482mmu-miR-126-3p160.49222.131.38<0.050.14183mmu-miR-15b56.1675.261.34<0.010.13499mmu-let-7i61.7881.151.31<0.050.14512
表3模型组较正常组表达下调的miRNAs
Table 3. The down-regulated miRNAs in model group compared with normal group
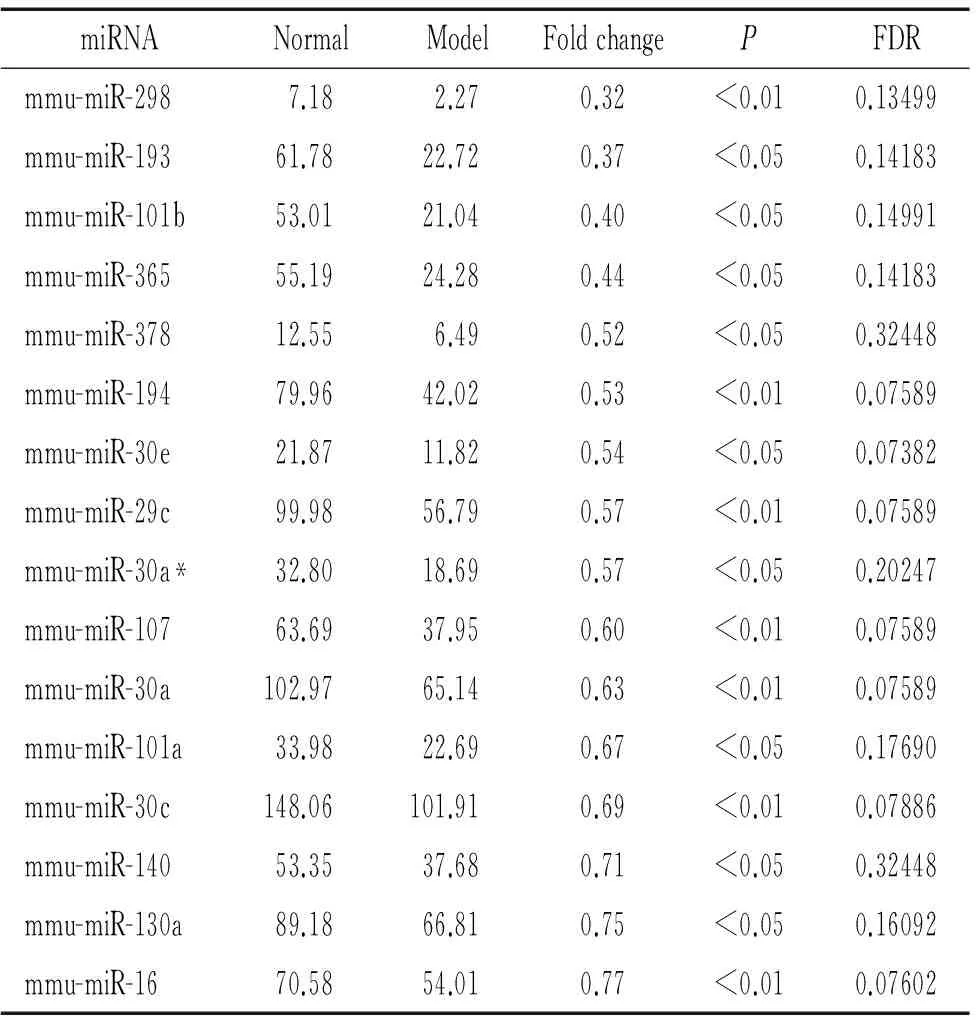
miRNANormalModelFoldchangePFDRmmu-miR-2987.182.270.32<0.010.13499mmu-miR-19361.7822.720.37<0.050.14183mmu-miR-101b53.0121.040.40<0.050.14991mmu-miR-36555.1924.280.44<0.050.14183mmu-miR-37812.556.490.52<0.050.32448mmu-miR-19479.9642.020.53<0.010.07589mmu-miR-30e21.8711.820.54<0.050.07382mmu-miR-29c99.9856.790.57<0.010.07589mmu-miR-30a*32.8018.690.57<0.050.20247mmu-miR-10763.6937.950.60<0.010.07589mmu-miR-30a102.9765.140.63<0.010.07589mmu-miR-101a33.9822.690.67<0.050.17690mmu-miR-30c148.06101.910.69<0.010.07886mmu-miR-14053.3537.680.71<0.050.32448mmu-miR-130a89.1866.810.75<0.050.16092mmu-miR-1670.5854.010.77<0.010.07602
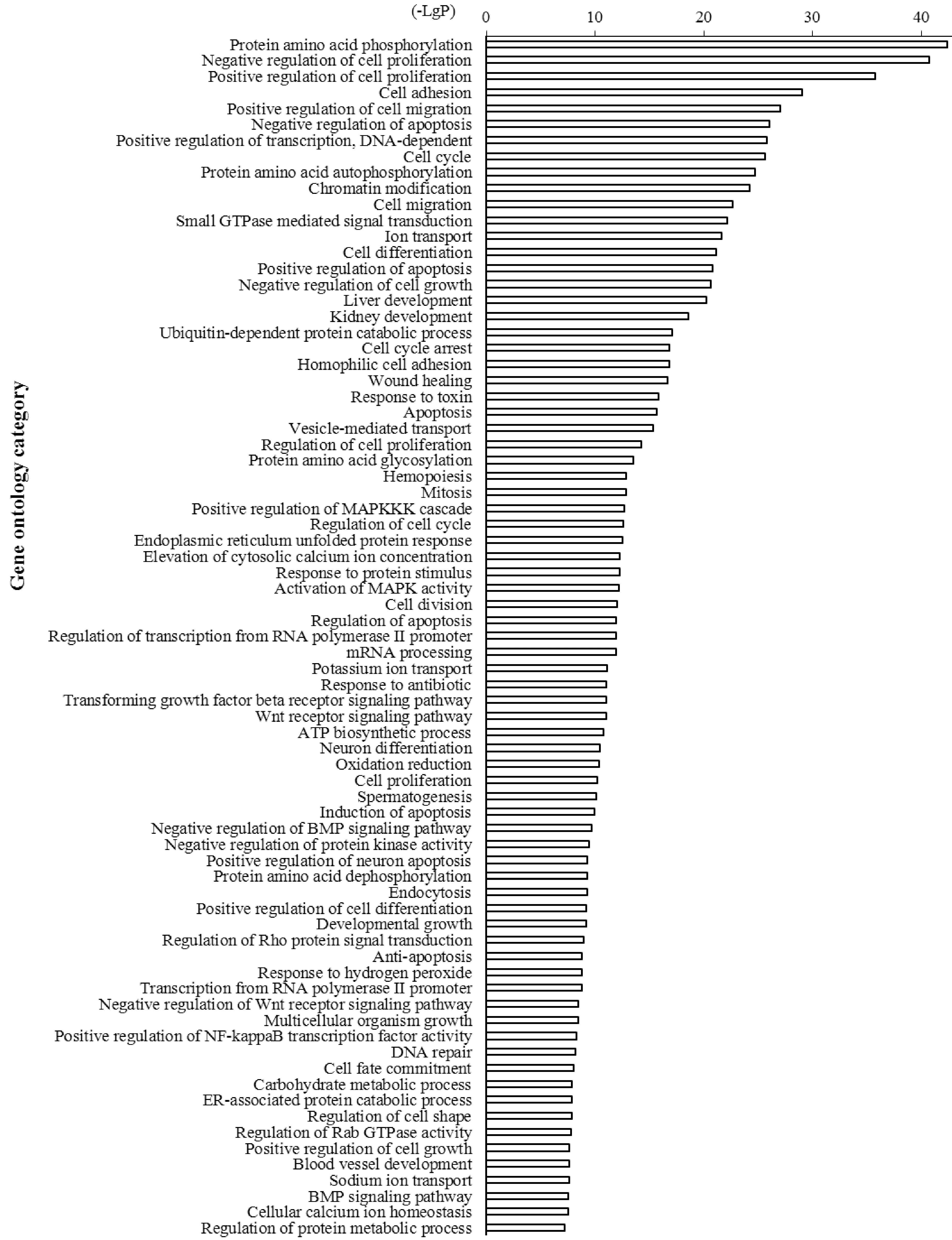
Figure 3. Gene ontology (GO) analysis based on target genes of the up-regulated miRNAs in model group compared with normal group. The vertical axis is the GO category, and the horizontal axis is the enrichment of GO.
图3模型组较正常组表达上调的miRNAs的靶基因GO分析
由显著性GO与基因的关系及miRNA与基因的关系,得到miRNA与显著性GO之间的关系,并对miRNA及GO在网络调控中的地位进行评价,评价标准以miRNA所调控的GO的个数(反之亦然)来衡量。结果发现关键的上调miRNA包括mmu-miR-322、mmu-miR-15b、mmu-miR-195、mmu-miR-200b、mmu-miR-214、mmu-miR-27a和mmu-miR-106b;关键的下调miRNA主要包括mmu-miR-16、mmu-miR-130a、mmu-miR-101b、mmu-miR-30a、mmu-miR-30e、mmu-miR-30c、mmu-miR-101a和mmu-miR-140。
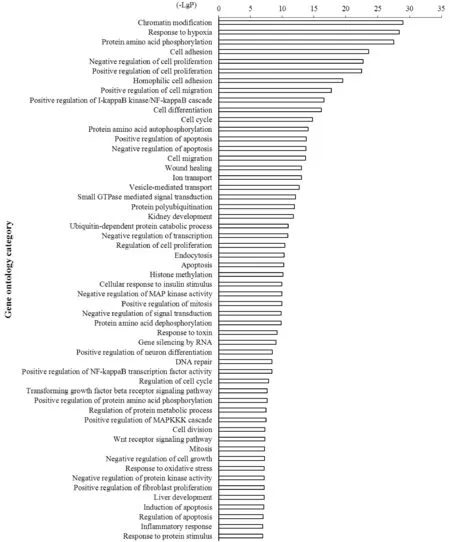
Figure 4. Gene ontology (GO) analysis based on target genes of the down-regulated miRNAs in model group compared with normal group. The vertical axis is the GO category, and the horizontal axis is the enrichment of GO.
图4模型组较正常组表达下调的miRNAs的靶基因GO分析
5差异miRNAs靶基因参与的信号转导通路分析(Pathway分析)
模型组较正常组上调的miRNAs的靶基因参与的显著性信号转导通路44条(图5),包括癌症相关通路、MAPK信号通路、黏着斑、内吞作用、骨架蛋白的调节、细胞周期、转化生长因子β(transforming growth factor β,TGF-β)信号通路等;而下调靶基因参与的显著性信号转导通路18条(图6),包括癌症相关通路、黏着斑、泛素调节的蛋白质水解作用、MAPK信号通路、细胞骨架的调节等。
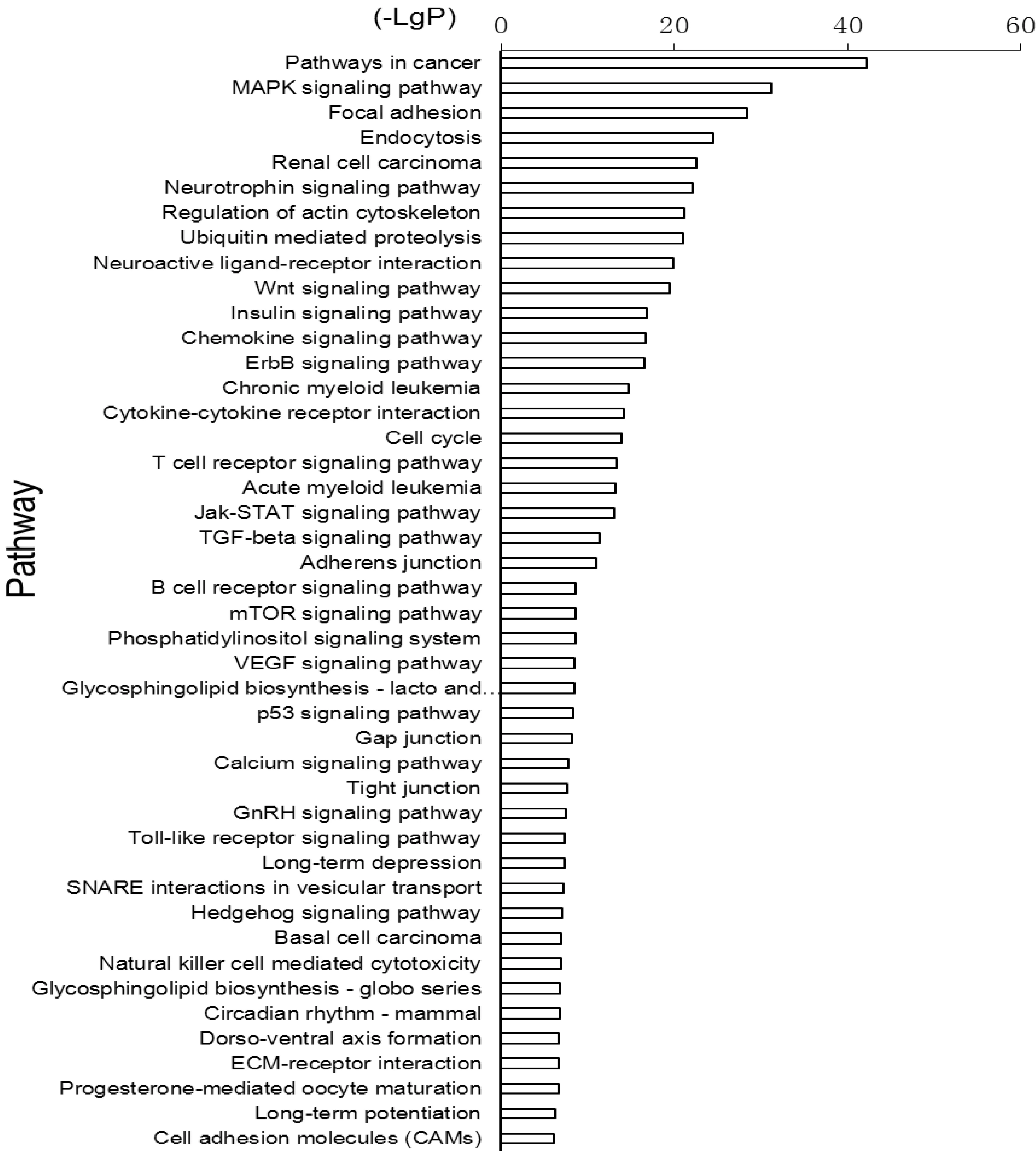
Figure 5. Pathway analysis based on target genes of the up-regulated miRNAs in model group compared with normal group. The vertical axis is the pathway category, and the horizontal axis is the enrichment of pathways.
图5模型组较正常组表达上调的miRNAs的靶基因Pathway分析
6差异miRNAs与交集靶基因的调控网络分析
通过靶基因的功能显著性分析和信号通路的显著性分析,得到显著性GO和Pathway及其所属的基因,取显著性GO所属靶基因与显著性Pathway所属靶基因的交集,利用miRNA与靶基因之间的靶向调控关系,构建miRNA-基因网络图(图7、8)。利用中心度来评价miRNA与基因在网络中的调控地位,发现,上调的关键miRNAs主要包括mmu-miR-200b、mmu-miR-322、 mmu-miR-106b、mmu-miR-23a、mmu-miR-15b、mmu-miR-195和mmu-miR-27a,其调控的关键靶基因主要包括Fut9、Acvr1c、Tbl1xr1、Acvr2a、Adrb2、Akt3及Smad7(图7);下调的关键miRNAs主要包括mmu-miR-16、mmu-miR-30e、 mmu-miR-30c、mmu-miR-30a、mmu-miR-130a、mmu-miR-101b、mmu-miR-101a及mmu-miR-107,其调控的关键靶基因主要包括Abl1、Rap1b、Runx1、Acvr1、Cul2、Cul4b及Map3k2(图8)。
讨 论
本研究采用miRNA芯片研究CCl4诱导的小鼠纤维化肝脏差异miRNA表达特点并分析其功能。通过比较小鼠纤维化肝脏与正常对照组的miRNA表达谱,共筛选出39个差异miRNAs,其中上调的23个,下调的16个。采用GO分析及KEGG 信号转导通路分析对差异miRNAs的功能进行了进一步探讨。

Figure 6. Pathway analysis based on target genes of the down-regulated miRNAs in model group compared with normal group. The vertical axis is the pathway category, and the horizontal axis is the enrichment of pathways.
图6模型组较正常组表达下调的miRNAs的靶基因Pathway分析
GO数据库是基因本体联合会(Gene Ontology Consortium)所建立的数据库,是一个跨物种、综合性、描述性的数据库[13],其目的在于建立一个适用于各种物种的、对基因和蛋白质功能进行限定和描述的、并能随着研究不断深入而更新的语义词汇标准。GO数据库用树状分层的方式对基因及蛋白功能进行详细的描述,阐明了基因功能之间的层次关系。功能层级越高,其定义的功能描述越笼统,范围越广;功能层级越低,其定义的功能描述越详细,范围越窄。GO数据库能帮助我们更好地理解基因功能之间的关系。基于GO数据库筛选出代表目标基因群显著的、准确的、靶向的基因功能的方法就称为GO分析[15]。其价值在于发现目标基因所带性状的最重要功能,发现同一基因在这种性状里发挥的主要功能或者非主要功能及判断研究目标在更大量的样本下准确与否。
对本实验筛选的差异miRNAs进行GO分析,发现模型组较正常组上调的miRNAs所对应的靶基因共参与80项显著性功能,包括蛋白氨基酸磷酸化作用、对细胞增殖的负向/正向调控、细胞黏附、对细胞移行的正向调控、对凋亡的负向调控、细胞周期、肝脏发育等;下调的miRNAs的靶基因共参与54项显著性功能,包括染色质改变、低氧应答、蛋白氨基酸磷酸化、细胞黏附等。进一步对miRNAs及GO在网络调控中的地位进行评价,评价标准以miRNA所调控的GO的个数(反之亦然)来衡量,发现关键的上调miRNA包括mmu-miR-322、mmu-miR-15b、mmu-miR-195、mu-miR-200b、mmu-miR-214、mmu-miR-27a和mmu-miR-106b;关键的下调miRNA主要包括mmu-miR-16、mmu-miR-130a、mmu-miR-101b、mmu-miR-30a、mmu-miR-30e、mmu-miR-30c、mmu-miR-101a和mmu-miR-140。
KEGG(Kyoto Encyclopedia of Genes and Genomes)是京都基因与基因组大百科全书的缩写,是系统分析基因(及其编码产物)间关系、基因功能、基因组信息的数据库,它有助于研究者把基因及表达信息作为一个整体网络进行研究[16]。基因组信息存储在GENES数据库里,包括完整和部分测序的基因组序列;更高级的功能信息存储在PATHWAY数据库里,包括图解的细胞生化过程如代谢、膜转运、信号传递、细胞周期,还包括同系保守的子通路等信息。KEGG提供的整合代谢途径查询十分出色,包括碳水化合物、核苷、氨基酸等的代谢及有机物的生物降解,不仅提供了所有可能的代谢途径,而且对催化各步反应的酶进行了全面的注解。KEGG是进行生物体内代谢分析和代谢网络研究的强有力工具。目前KEGG Pathway分为8个类别,分别为总体网络、代谢过程、遗传信息传递、环境信息传递、胞内生物过程、生物体系统、人类疾病及药物研发。

Figure 7. Analysis of miRNA-gene (mRNA) network based on the up-regulated miRNAs in model group compared with normal group. Boxes represent miRNA, and cycle nodes represent mRNA. Lines show the inhibitory effect of miRNA on mRNA.
图7模型组较正常组表达上调的miRNAs的miRNA-基因网络图
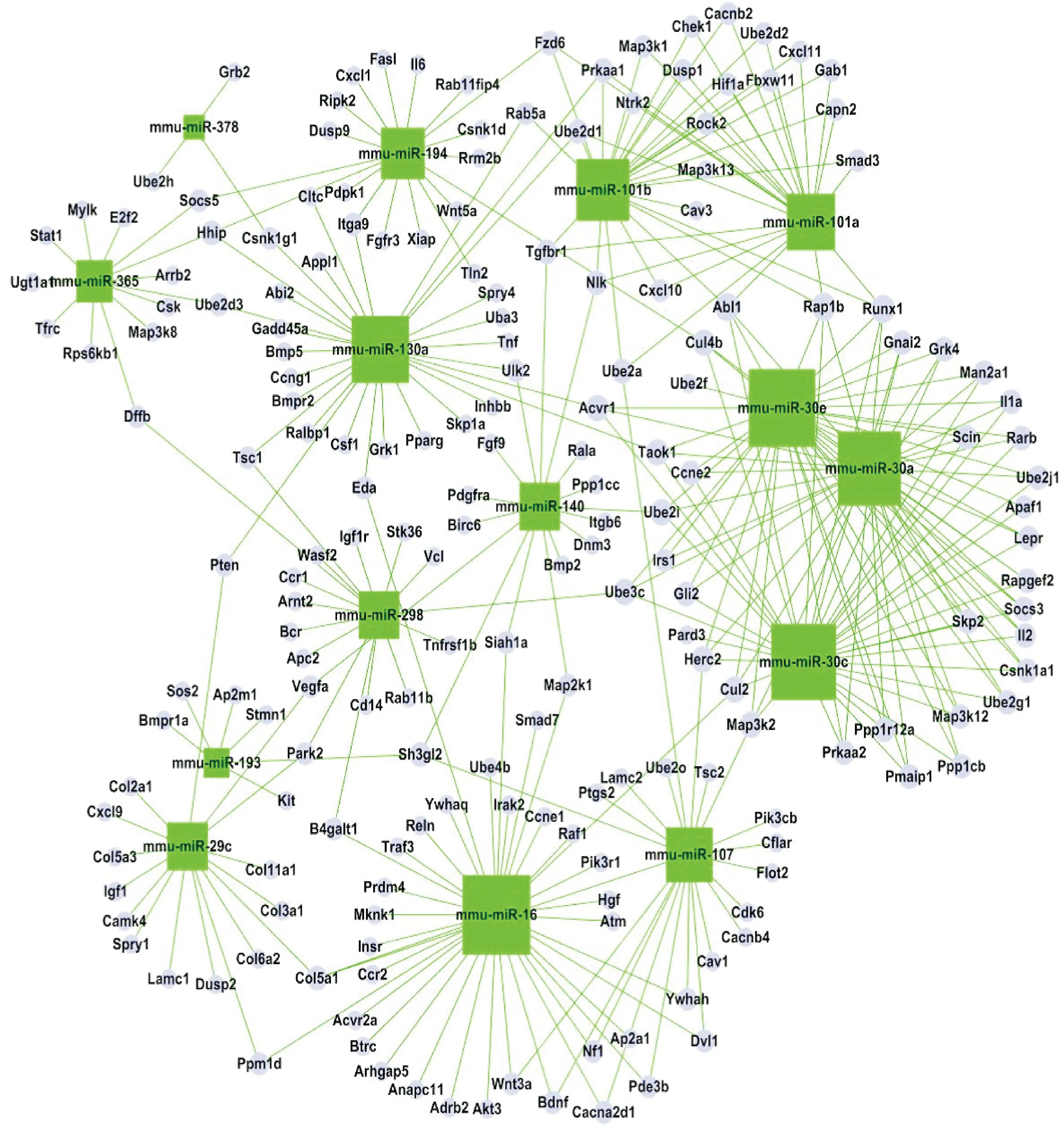
Figure 8. Analysis of miRNA-gene (mRNA) network based on the down-regulated miRNAs in model group compared with normal group. Boxes represent miRNA, and cycle nodes represent mRNA. Lines show the inhibitory effect of miRNA on mRNA.
图8模型组较正常组表达下调的miRNAs的miRNA-基因网络图
基于KEGG数据库,对本实验筛选的差异miRNAs对应的靶基因进行信号转导通路分析,发现上调的miRNAs的靶基因参与的显著性信号转导通路44条,包括癌症相关通路、MAPK信号通路、黏着斑、内吞作用、骨架蛋白的调节、细胞周期、TGF-β信号通路、VEGF信号通路等;而下调靶基因参与的显著性信号转导通路18条,包括癌症相关通路、黏着斑、泛素调节的蛋白质水解作用、MAPK信号通路、细胞骨架的调节、细胞与细胞因子受体关联作用、趋化因子信号通路等。
对显著性GO所属靶基因与显著性Pathway所属的靶基因取交集,利用图论的方法对网络中miRNA和基因在网络中的调控地位进行评价,评价的标准以miRNA和基因在网络中的度来衡量,度表示网络中的miRNA调控靶基因的数目(反之亦然)[17]。结果发现关键的上调miRNAs包括mmu-miR-200b、mmu-miR-322、mmu-miR-106b、mmu-miR-23a、mmu-miR-15b、mmu-miR-195和mmu-miR-27a;上调miRNA调控的关键靶基因主要包括Acvr1c、Tbl1xr1、cvr2a、Adrb2、Akt3、Slc8a1、Smad7等。其中Smad7是TGF-β1/Smads信号转导通路的负性调节因子,可抑制胞内R-Smad磷酸化及与Smad4的结合,促进受体的降解[19-21]。细胞内关键的下调miRNA主要包括mmu-miR-16、mmu-miR-30e、mmu-miR-30c、mmu-miR-30a、mmu-miR-130a、mmu-miR-101b、mmu-miR-101a和mmu-miR-107;下调miRNA调控的关键靶基因主要包括Abl1、Rap1b、Runx1、Acvr1、Cul2、Cul4b、Herc2和Map3k2。
综上所述,肝纤维化形成的各个环节,包括细胞的增殖与活化、细胞凋亡、细胞黏附、细胞迁移、Wnt受体信号转导通路、TGF-β1/Smads信号转导通路等都可能受miRNAs的调控。
[1] Lee RC, Feinbaum RL, Ambros V. TheC.elegansheterochronic genelin-4 encodes small RNAs with antisense complementarity tolin-14[J]. Cell, 1993,75(5):843-854.
[2] Zhang B, Pan X, Anderson TA. MicroRNA: a new player in stem cells[J]. J Cell Physiol, 2006, 209(2):266-269.
[3] Tanno B, Cesi V, Vitali R, et al. Silencing of endogenous IGFBP-5 by microRNA interference affects proliferation, apoptosis and differentiation of neuroblastoma cells[J]. Cell Death Differ, 2005, 12(3):213-223.
[4] Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease[J]. Development, 2005, 132(21):4653-4662.
[5] 许蜜蝶, 李微卿, 余宏宇. Micro RNA与肝病及肝纤维化相关研究 [J].中华病理学杂志,2011,40(1):65-67.
[6] 张 帅, 李有杰, 张 超, 等. miRNA在去甲斑蝥素致K562细胞凋亡中的作用研究[J].中国病理生理杂志, 2011, 27(3): 499-503.
[7] Pogribny IP, Starlard-Davenport A, Tryndyak VP, et al. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice[J]. Lab Invest, 2010, 90(10):1437-1446.
[8] Tsukamoto H, Matsuoka M, French SW. Experimental models of hepatic fibrosis: a review[J]. Semin Liver Dis, 1990, 10(1):56-65.
[9] 中华肝脏病学会肝纤维化学组. 肝纤维化诊断及疗效评估共识[J]. 中华肝脏病杂志, 2002, 10(5):327-328.
[10] Jamall IS, Finelli VN, Que Hee SS. A simple method to determine nanogram levels of 4-hydroxyproline in biological tissues[J]. Anal Biochem, 1981, 112(1):70-75.
[11] Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments[J]. Bioinformatics,2003, 19(18):2448-2455.
[12] Yang H, Crawford N, Lukes L, et al. Metastasis predictive signature profiles pre-exist in normal tissues[J]. Clin Exp Metastasis,2005, 22(7):593-603.
[13] Ashburner M, Ball CA, Blake JA, et al. Gene ontology:tool for the unification of biology. The Gene Ontology Consortium[J]. Nat Genet, 2000, 25(1):25-29.
[14] Dupuy D, Bertin N, Hidalgo CA, et al. Genome-scale analysis ofinvivospatiotemporal promoter activity inCaenorhabditiselegans[J]. Nat Biotechnol,2007, 25(6):663-668.
[15] Yi M, Horton JD, Cohen JC, et al. WholePathwayScope: a comprehensive pathway-based analysis tool for high-throughput data[J]. BMC Bioinformatics, 2006, 7:30.
[16] Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome[J]. Nucleic Acids Res, 2004, 32(Database issue):D277-D280.
[17] Joung JG, Hwang KB, Nam JW, et al. Discovery of microRNA-mRNA modules via population-based probabilistic learning[J]. Bioinformatics, 2007, 23(9):1141-1147.
[18] Shalgi R, Lieber D, Oren M, et al. Global and local architecture of the mammalian microRNA-transcription factor regulatory network[J]. PLoS Comput Biol, 2007, 3(7):e131.
[19] Liu C, Gaca MD, Swenson ES, et al. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent[J]. J Biol Chem,2003,278(13):11721-11728.
[20] Kawabata M, Miyazono K. Signal transduction of the TGF-beta superfamily by Smad proteins[J]. J Biochem,1999,125(1):9-16.
[21] Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily[J]. Science,2002,296(5573):1646-1647.
FunctionalanalysisofdifferentialmicroRNAexpressionprofileinmousefibroticlivertissuesinducedbyCCl4
WANG Qing-lan1,2, LI Jun-xia1, LÜ Jing1, TAO Yan-yan1, YAN Xiu-chuan1, LIU Cheng-hai1,2,3,4
(1InstituteofLiverDiseases,ShuguangHospitalAffiliatedtoShanghaiUniversityofTraditionalChineseMedicine,2ShanghaiClinicalKeyLaboratoryofTraditionalChineseMedicine,3KeyLaboratoryofLiverandKidneyDiseases,MinistryofEducation,ShanghaiUniversityofTraditionalChineseMedicine,4E-InstituteofTCMInternalMedicine,ShanghaiMunicipalEducationCommission,Shanghai201203,China.E-mail:chenghailiu@hotmail.com)
AIM: To discover the expression profile of microRNAs (miRNAs) in mouse fibrotic liver tissues induced by carbon tetrachloride (CCl4), and to investigate the functions of these differential miRNAs based on the gene ontology (GO) analysis and KEGG Pathway analysis.METHODSThe mice were randomly divided into normal group and model group. Liver fibrosis was induced by subcutaneous injection of CCl4. miRNA expression profile of the liver tissues was assayed by a mouse miRNA microarray (Agilent 12.0). The differential expression of miRNAs between the normal and model mice was screened, and GO analysis and KEGG Pathway analysis were performed to determine the functions of these differential miRNAs.RESULTSThirty-nine miRNAs with differential expression were discovered in the model mice compared with the normal mice, among which 23 were up-regulated and 16 were down-regulated. GO analysis and KEGG Pathway analysis indicated that most pathological processes of liver fibrosis regulated by miRNAs included cell proliferation and activation, cell apoptosis, cell cycle, cell adhesion, inflammatory reaction, cell migration, transforming growth factor β (TGF-β) signaling pathway, Wnt signaling pathway and proteometabolism process. GO analysis revealed that the key up-regulated miRNAs were mmu-miR-322, mmu-miR-15b, mmu-miR-195, mmu-miR-200b and mmu-miR-214, and the key down-regulated miRNAs were mmu-miR-16, mmu-miR-130a, mmu-miR-101b, mmu-miR-30a and mmu-miR-30e. Analyzing the target genes screened out by GO analysis and Pathway analysis simultaneously, we found that the key up-regulated miRNAs included mmu-miR-200b, mmu-miR-322, mmu-miR-106b, mmu-miR-23a and mmu-miR-15b, and the key down-regulated miRNAs included mmu-miR-16, mmu-miR-30e, mmu-miR-30c, mmu-miR-30a and mmu-miR-130a.CONCLUSIONDifferential expression of miRNAs is discovered in mouse fibrotic liver tissues induced by CCl4compared with the normal liver tissues. Most of the pathological processes involved in liver fibrosis may be regulated by miRNA, such as cell proliferation and activation, cell adhesion and apoptosis, cell migration and differentiation, metabolism, TGF-β receptor signaling pathway and so on.
MicroRNA; Liver fibrosis; Differential expression; Carbon tetrachloride
R363.2
A
10.3969/j.issn.1000- 4718.2013.12.014
1000- 4718(2013)12- 2201- 11
2013- 01- 11
2013- 11- 08
国家自然科学基金资助项目(No.30901943;No.81270053);上海高校创新团队建设项目(第一期);国家中医药管理局中医肝胆病重点学科(No.2010sh)
△通讯作者Tel: 021-20256523; E-mail: chenghailiu@hotmail.com
