力竭运动对小鼠骨骼肌细胞自噬的影响及相关调节机制研究
2013-10-18祖靓,朱荣
祖 靓,朱 荣
大自噬(统称“细胞自噬”)是从酵母菌到人类在进化上高度保守的细胞内分解代谢过程。长寿蛋白质或衰老、损伤的细胞器被自噬小体包裹后转运至溶酶体降解,部分产物转变成物质代谢和能量代谢循环的底物,可以满足细胞在饥饿等应激状态下的能量需求。骨骼肌大约占体重的40%,是哺乳动物主要的蛋白质储存库,在能量耗竭时是氨基酸的主要来源,同时还是机体主要的动力器官。多项研究表明,维持骨骼肌的完整性需要适宜的自噬水平。过高或过低的自噬都不利于骨骼肌的健康,自噬过高引起骨骼肌质量下降,自噬过低又引起骨骼肌纤维降解和肌无力[24]。在敲除自噬基因ATG 7的小鼠骨骼肌中观察到肌肉萎缩和肌力下降[18]。在自然衰老的动物骨骼肌中发现细胞自噬关键蛋白LC3-II、p62表达增高,去神经支配的年轻动物中自噬蛋白表达也显著增加[21]。自噬缺陷还会降低小鼠的运动耐力,引起糖代谢异常[11]。以上研究均表明,细胞自噬在维持骨骼肌内环境稳态中的作用至关重要。2012年Garber[7]在Science杂志上指出,运动训练对机体代谢的良性效应可利用自噬现象来解释,使细胞自噬成为运动科学领域的研究热点。然而,运动训练对骨骼肌内细胞自噬影响的研究刚刚起步,调节机制也未完全明确。
腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)是真核细胞内重要的能量感受器,对ATP/AMP变化非常敏感,被誉为能量平衡的中心调节因子。体外实验发现,HEK293细胞在葡萄糖缺乏时,AMPK可以直接与自噬启动早期阶段具有重要作用的丝氨酸/苏氨酸蛋白激酶——失调51样激酶-1(uncoordinated 51like kinase-1,ULK1)结合,并磷酸化ULK1Ser317和Ser777位点,提高ULK1酶活性,促进细胞自噬[15]。本文拟观察力竭跑台运动对小鼠骨骼肌细胞自噬相关蛋白表达的影响,并试图研究AMPK在活体骨骼肌细胞自噬中的作用和机制。
1 实验材料与方法
1.1 实验对象
清洁级健康雄性C57BL/6小鼠96只,2月龄,体质量19.5±1.4g,由温州医科大学实验动物中心提供,自由进食水,饲养环境为20~25℃,湿度45%~60%,12h/12h昼夜节律光照。
1.2 实验分组
适应性饲养1周后,根据体重随机将96只小鼠分为AICAR注射组(AICAR Injection,AI)、力竭运动组(Exhaustive Exercise,EE)、Compound C 注射+力竭运动组(Compound C+Exhaustive Exercise,CE)和相应的安静对照组(Sedentary Control,SC)。根据取材时间,AI组又分注射后1h,2h和6h组。EE,CE组分别分为运动后即刻和注射后即刻、3h、6h、12h和24h组,加上各自对照组总共16组,每组6只。SC组笼中常规饲养,不进行训练;CE组及其对照组在运动开始前20min按照0.5g/kg体重(预实验确定)大腿外侧皮下分别注射AMPK活性抑制剂Compound C(Merk)和等剂量生理盐水,EE组及其对照组均注射等剂量生理盐水;AI组及其对照组分别按照0.7g/kg体重[8]皮下注射 AMPK激活剂 AICAR(Santa Cruz)和等剂量生理盐水。
1.3 动物运动方案及取材
在正式实验前,运动组小鼠先进行2~3次适应性跑台练习,每次先以5m/min的强度进行3min热身运动,然后逐渐加速至11m/min,第一次持续10min,第二次持续30min,第三次持续50min。隔天进行正式实验。运动组小鼠进行速度为11m/min(75%˙VO2max)、坡度为0°跑台运动[6],直至力竭。力竭标准:当小鼠不能维持跑台预定速度,随转动皮带拖至跑台后端的挡板(泡沫自制)处停留达30s以上,毛刷驱赶或手轻推均无效,视为力竭[19]。采用颈椎脱臼法处死小鼠,在30~60s内采集双侧后肢腓肠肌标本,包裹在锡纸中,液氮骤冷,转移至-80℃超低温冰箱(Thermo)保存备用。
1.4 ELISA法测定AMPK酶活性
40mg腓肠肌于蛋白裂解液中充分匀浆,12000rpm离心30min,取上清液,考马斯亮蓝法测蛋白浓度。PBS稀释样品至20μg/ml,根据上海蓝基生物ELISA试剂盒说明书(产品编号:E02A0043)向96孔酶标板上加入50μl浓度依次稀释的样品(最低不得低于2μg/ml)。塑料膜封板室温孵育2h后,200μl PBS清洗酶标板。200μl封闭液(PBS稀释的5%脱脂牛奶)封闭,塑料膜封板4℃过夜。次日,200μl PBS清洗酶标板两次,每孔加入100μl一抗稀释液,塑料膜封板室温孵育2h。PBS清洗酶标板,每孔加入100μl二抗(封闭液稀释),塑料膜封板室温孵育1.5 h。PBS清洗酶标板,每孔加入50μl HRP发光液,5min后加入100μl反应终止液,通过酶标仪(Bio-rad,美国)在450nm波长下测定各孔OD值。以标准品浓度为横坐标,OD值为纵坐标,绘制标准曲线。根据回归方程OD=浓度×0.133(U/L),计算出待测样品AMPK浓度。用AMPK浓度/总蛋白浓度比值表示AMPK活性。
1.5 Western blot法测定骨骼肌相关蛋白表达
将上样缓冲液与上述骨骼肌蛋白样品等体积混合煮沸10min。在垂直电泳仪上用相同体积的15μg蛋白质样品分别经6%、15%、10%SDS-PAGE分离p-ULK1、LC3、ATG 7、Beclin1、p62和β-tubulin后,转移于PVDF膜上。1∶1000兔抗鼠一抗4℃静置孵育过夜,PBS洗涤3次,再以1∶1000辣根过氧化物酶标记的羊抗兔IgG(H+L)二抗室温孵育1h,PBS充分洗涤后,在暗室使用ECL试剂盒发光显影,X光胶片压片曝光。采用Champ Gel 2000凝胶系统分析软件,扫描定量各条带的相对灰度值,在与内参比较后,以对照组条带灰度值为100%,其它组条带灰度值与对照组条带灰度值的比值,即为其相对表达量(%)。
1.6 免疫共沉淀法测定AMPK和ULK1结合量
60mg新鲜腓肠肌组织,提取蛋白,并测定浓度(同上)。按照碧云天Protein A+G Agarose(产品编号P2012)试剂盒说明书,向待测样品加入1μg AMPK一抗,4℃缓慢摇动过夜(摇床置于冰箱内)。加入25μl Protein A+G,4℃缓慢摇动3h,捕获蛋白复合物。2500rpm(约1000 g)离心5min,吸除上清。500μl预冷PBS洗剂沉淀5次。最后一次洗剂后去除上清,加入30μl SDS-PAGE上样缓冲液,瞬时离心。沸水浴处理5min,样品用8%SDSPAGE分离AMPK和ULK1。接下来的步骤同Western blot,AMPK和ULK1蛋白以积分灰度值的比值表示。
1.7 数据统计
采用SPSS 13.0统计软件对所获得的实验数据进行统计分析,所有数据以平均数±标准差表示。组间比较采用单因素方差分析。以P<0.05为显著性差异标准,以P<0.01为非常显著性差异标准。
2 结果
2.1 AICAR和力竭运动对骨骼肌AMPK活性的影响
小鼠腓肠肌AMPK活性在AICAR注射后1h时显著升高(P<0.01),2h后仍显著高于对照组(P<0.05),但与1h组相比,下降明显(P<0.01),6h时基本恢复(图1A);力竭运动后即刻,AMPK活性明显升高(P<0.01),3h时显著下降,但仍显著高于对照组(P<0.05),6h时基本恢复;Compound C+力竭运动组,AMPK活性在运动后与对照组相比变化不大,但在运动后即刻显著低于力竭运动组(P<0.01,图1B)。
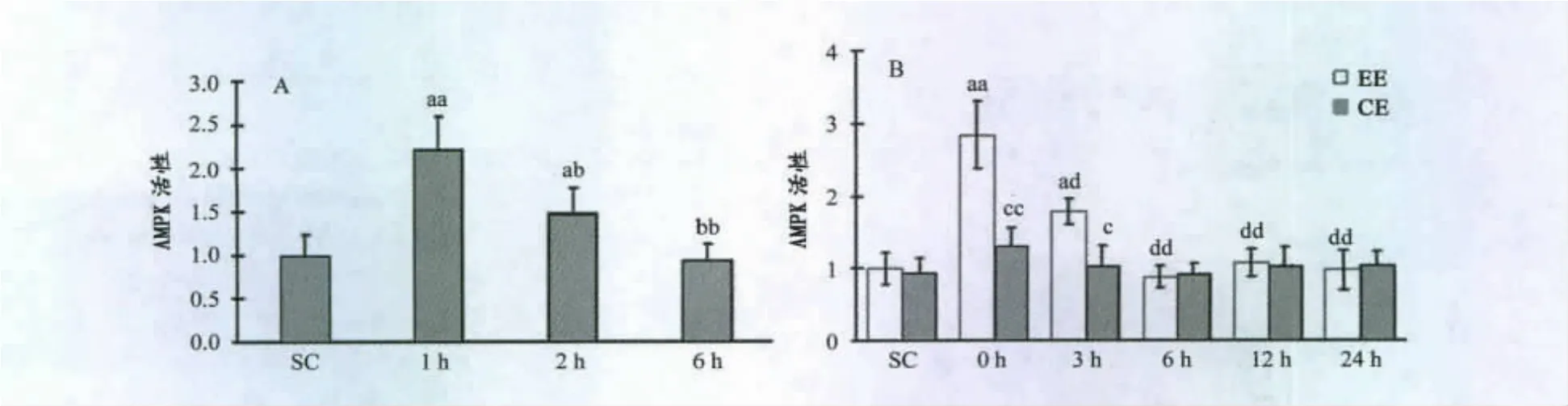
图1 本研究AICAR和力竭运动对小鼠骨骼肌AMPK活性的影响Figure 1.Effects of AICAR and Exhaustive Exercise on AMPK Activity in Murine Skeletal Muscle
2.2 AICAR对骨骼肌AMPK/ULK1的影响
如图2所示,AMPK/ULK1结合量在AICAR注射后1 h时显著增加(P<0.01),2h时明显下降(P<0.05),6h时基本恢复到正常水平。
2.3 AICAR对骨骼肌自噬关键蛋白ULK1、LC3等含量的影响
如图3、4、5所示,ULK1Ser317磷酸化水平在AICAR注射后1h时显著高于对照组(P<0.01),2h时基本下降到正常水平;与对照组相比,LC3-I在AICAR注射1h时显著下降(P<0.05),6h时基本恢复(P<0.05);LC3-II在AICAR注射后1h时显著增加(P<0.01),恢复期(2h,6h)显著下降(P<0.01);LC3-II/LC3-I比值在 AICAR注射后1h时显著升高(P<0.01),恢复期(2h,6h)显著下降(P<0.01)。p62在AICAR注射后有下降趋势,但没有统计学意义;Beclin1和Atg7变化趋势基本一致:AICAR注射后1h显著下降(P<0.05),2h时基本恢复到正常水平。

图2 本研究AICAR对小鼠骨骼肌AMPK/ULK1结合影响Figure 2.Effects of AICAR on Binding Activity of AMPK/ULK1in Murine Skeletal Muscle

图3 本研究AICAR对小鼠骨骼肌ULK1Ser317磷酸化水平的影响Figure 3.Effects of AICAR on Phosphorylation of ULK1Ser317 Site in Murine Skeletal Muscle

图4 本研究AICAR对小鼠骨骼肌LC3-I向LC3-II转化的影响Figure 4.Effects of AICAR on Conversion of LC3-I to LC3-II in Murine Skeletal Muscle
2.4 力竭运动对骨骼肌AMPK/ULK1结合的影响
图6表明,力竭运动组AMPK/ULK1结合量在运动后即刻显著高于对照组(P<0.01),3h时仍显著高于对照组(P<0.05),6h时基本恢复;与对照组相比,Compound C注射运动组AMPK/ULK1结合在运动后变化不明显,但与力竭运动组相比,Compound C注射+力竭运动组AMPK/ULK1结合在运动后即刻显著低于同一时间取材的力竭运动组 (P<0.01)。
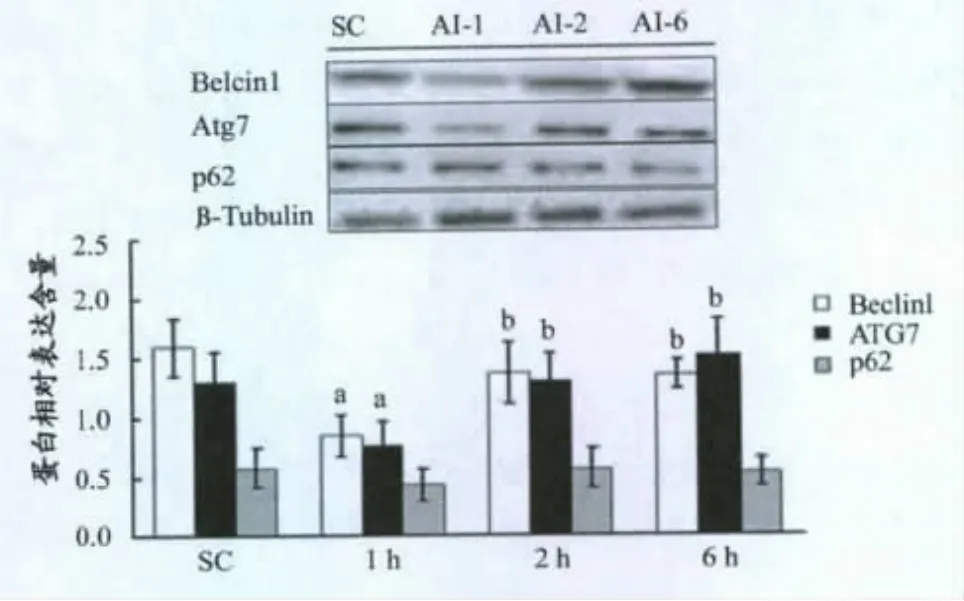
图5 本研究AICAR对小鼠骨骼肌Belcin1、Atg7、p62的影响Figure 5.Effects of AICAR on Beclin1,Atg7 and p62Expression in Murine Skeletal Muscle
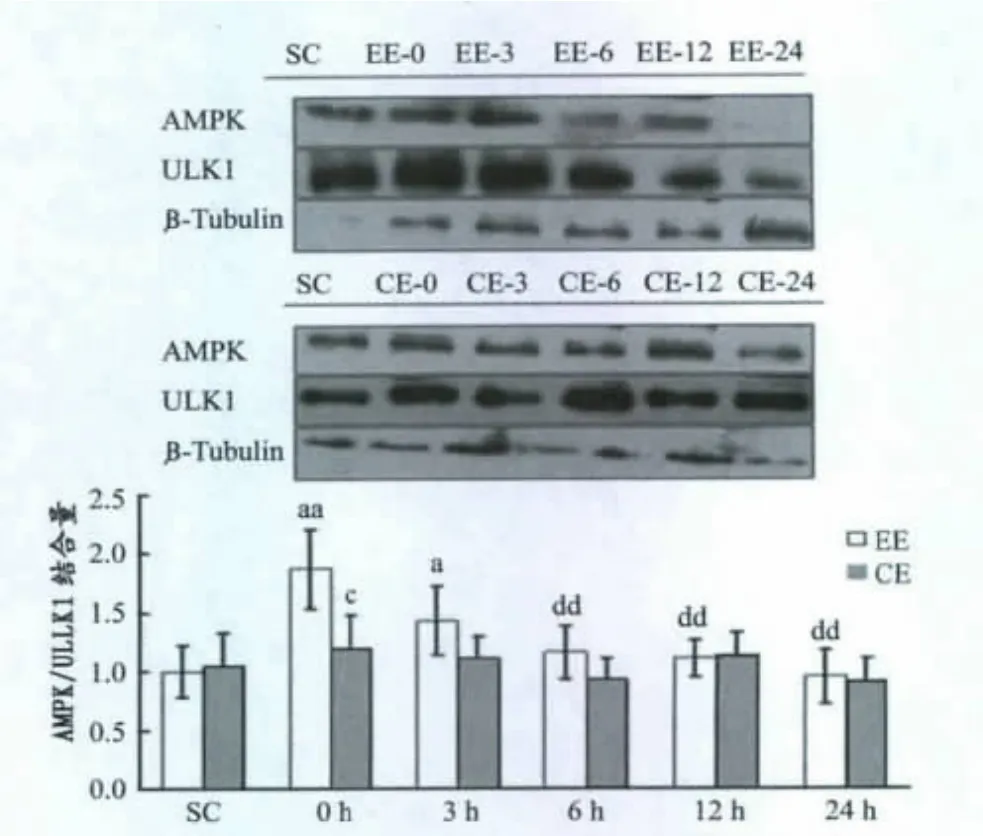
图6 本研究力竭运动对小鼠骨骼肌AMPK/ULK1结合的影响Figure 6.Effects of Exhaustive Exercise on Binding Activity of AMPK/ULK1in Murine Skeletal Muscle
2.5 力竭运动对骨骼肌自噬关键蛋白 ULK1、LC3、ATG7等的影响
由图7~12可知,力竭运动后即刻ULK1Ser317磷酸化水平显著提高(P<0.01),3h时仍显著高于对照组(P<0.01),12h时已基本恢复;Compound C注射组p-ULK1Ser317在运动结束后与对照组相比无显著性差异,但运动后即刻显著低于力竭运动组(P<0.05)。力竭运动组LC3-I在运动后即刻显著下降(P<0.05),6时基本恢复;LC3-II在运动后即刻显著升高(P<0.01),恢复期(6h,12h,24h)显著下降(P<0.01),12h时基本恢复;LC3-II/LC3-1比值 变化趋 势同 LC3-II;Compound C注射组LC3-I在运动后即刻也显著下降(P<0.05),但显著低于EE运动后即刻组(P<0.05),3h时基本恢复,LC3-II在运动后即刻显著升高(P<0.01),但低于EE运动后即刻组,恢复期(6h,12h,24h)显著下降,LC3-II/LC3-I比值变化趋势同LC3-II;Beclin1在力竭运动后即刻显著低于对照组(P<0.01),6h时已基本恢复,Compound C注射组Belcin1运动后即刻显著下降,但显著高于力竭运动后即刻组,3h时基本恢复;力竭运动组ATG7在运动后恢复期一直低于对照组,12h时尚未恢复,Compound C注射组ATG7在运动后恢复期显著低于对照组(P<0.05);p62在运动后即刻显著下降(P<0.05),3h时已经基本恢复,Compound C注射组p62运动后即刻显著增加(P<0.01),恢复期显著下降。

图7 本研究力竭运动对小鼠骨骼肌p-ULK1含量的影响Figure 7.Effects of Exhaustive Exercise on p-ULK1Level in Murine Skeletal Muscle
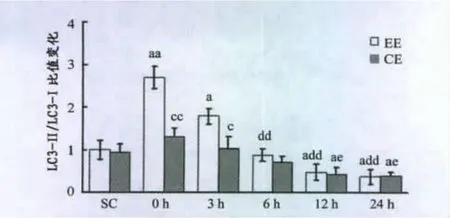
图9 本研究力竭运动对小鼠骨骼肌LC3-II/LC3-I比值的影响Figure 9.Effects of Exhaustive Exercise on Ratio of LC3-II/LC3-I in Murine Skeletal Muscle

图10 本研究力竭运动对小鼠骨骼肌Beclin1含量的影响Figure 10.Effects of Exhaustive Exercise on Beclin1Expression in Murine Skeletal Muscle
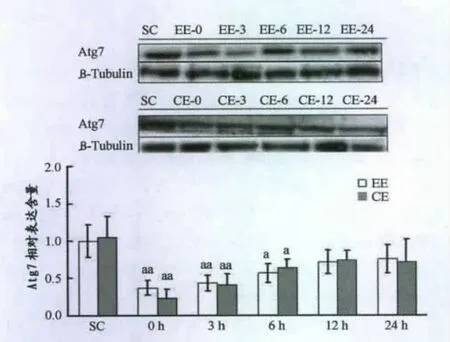
图11 本研究力竭运动对小鼠骨骼肌Atg7含量的影响Figure 11.Effects of Exhaustive Exercise on Atg7Expression in Murine Skeletal Muscle

图12 本研究力竭运动对小鼠骨骼肌p62含量的影响Figure 12.Effects of Exhaustive Exercise on p62Expression in Murine Skeletal Muscle
3 讨论
3.1 AICAR对骨骼肌细胞自噬的影响
自噬是真核生物体内广泛存在的应激应答机制[1],但骨骼肌内自噬上游调节机制尚不清楚。早期研究表明,ULK1是启动细胞自噬的重要调节因子。ULK1活性主要受磷酸化和去磷酸化修饰调控,目前已经确定了至少30个ULK1磷酸化位点[17],其中,mTOR和AMPK是目前已经证实的两个ULK1调节激酶。基础状态下,mTOR通过与ULK1结合磷酸化ULK1及其结合蛋白Atg13,降低Atg13和ULK1之间亲和力,抑制ULK1酶活性,抑制自噬[13]。饥饿状态下,AMPK成为促进自噬的主要调节因子。在AICAR处理的小鼠胚胎成纤维细胞 (Mouse Embryonic Fibroblast)中,Lee 等[17]发 现,AMPK磷酸化mTOR结合蛋白—Raptor,抑制mTORC1复合物活性,解除其自噬抑制效应,启动自噬。该研究还发现,AMPK与ULK1蛋白脯氨酸-丝氨酸富集结构域(proline-serine rich domain,PS)内的654-838氨基酸序列结合对ULK1介导的自噬激活是不可或缺的。Sanchez等[23]在C2C12成肌细胞和C2C12肌管中也发现AICAR处理后AMPK与ULK1结合显著增多,GFP-LC3标记的自噬小体数量增多。AMPK与ULK1的结合引起何种生理效应尚不清楚。Kim等[15]发现,葡萄糖缺乏时,ULK1激酶活性升高,主要表现为自身磷酸化水平升高,Compound C处理或者AMPKα缺失后这种变化被抑制,表明葡萄糖缺乏诱导的ULK1磷酸化激活是AMPK依赖性的,他们还发现,AMPK可以直接磷酸化ULK1Ser317和Ser777位点,促进自噬。在体骨骼肌中是否存在AMPK对ULK1及其介导的自噬调控尚不清楚。
张国华等[2]通过 AICAR(0.5g/kg体重)注射观察大鼠腓肠肌AMPK活性变化,发现AICAR注射1h后AMPK活性升高62%,2h时已基本恢复到正常水平。本研究发现,腓肠肌 AMPK活性在AICAR(0.7g/kg体重)注射后1h时显著升高120%(P<0.01),恢复期(2 h,6h)AMPK 活性显著下降(P<0.05),但是,在2h时仍显著高于对照组(P<0.05),可能是由于剂量不同造成的,也可能是小鼠与大鼠之间存在个体差异。骨骼肌ULK1Ser317磷酸化水平在AICAR注射后1h时显著提高,表明AICAR在激活AMPK的同时显著提高ULK1 Ser317磷酸化水平(P<0.01)。同时本实验还发现,AMPK与ULK1结合在AICAR注射后的变化与ULK1磷酸化水平变化趋势基本一致,提示AMPK结合并磷酸化ULK1,提高其酶活性。
LC3蛋白翻译后不久,就被胞浆中Atg4酶切,转变为LC3-I形式,在Atg7和Atg3作用下,LC3-I与磷脂酰乙醇胺(phosphatidylethanolamine,PE)结合,下形成具有膜结合能力的LC3-II,为检测自噬的标志性蛋白[3]。Ge等[9]在心肌细胞内发现 AICAR(500μM)处理50min后LC3-II表达显著升高,LC3-II/LC3-I比值变大,但对LC3-I表达并没有影响。本研究发现,AICAR注射后1h时LC3-II含量明显增加,LC3-I下降,表明LC3-I向LC3-II转化增多,而LC3-II/LC3-I比值增大进一步表明,此时AICAR显著提高安静状态下骨骼肌的自噬水平,这与Ge[9]的研究结果基本一致。2h时LC3-I仍显著低于对照组,LC3-II表达较AI1h组显著下降(P<0.01),LC3-II/LC3-I比例也显著下降,表明自噬水平在AICAR注射后1h达到顶峰,2h时已呈下降趋势,LC3-II经自噬-溶酶体途径降解,这也与JU等[14]研究发现安静状态下,自噬通路启动后LC3-II水平有可能升高、下降、甚至不变的结论是一致的。本实验首次在活体骨骼肌中验证了AICAR注射诱导的AMPK激活促进了安静状态下细胞自噬活化。
自噬启动后,整个过程还有一些关键蛋白的参与,包括Beclin1、Atg7、p62等。Beclin1 与 PI3KIII(class III phosphatidyl-inositol 3kinase),Vps34(vacuolar protein sorting 34)形成的复合物是细胞自噬囊泡成核(vesicle nucleation)阶段最重要的调节因子,与自噬小体膜的合成关系密切[4]。紧接着的囊泡伸展(vesicle elongation)阶段,E1泛素样酶Atg7在Atg12-Atg5泛素样复合物反应中具有重要作用,Atg7介导的这一反应还能促进PE与LC3结合以及LC3-I向LC3-II转化[22]。自噬的主要生理作用是降解胞浆中堆积的衰老蛋白,p62是自噬过程中底物识别的重要配体蛋白。自噬激活时,p62通过自身含有的LC3相互作用区(LC3interaction region,LIR)以配体作用方式与LC3结合,同时又通过自身泛素结合区(ubiquitin binding area,UBA)与泛素化蛋白或细胞器结合,将底物运至自噬小体,最终进入溶酶体途径降解。Egan等[5]发现,在AMPK和ULK1基因缺乏的小鼠肝脏以及原代肝细胞中,p62堆积,线粒体自噬过程受阻,因此常常将p62作为自噬过程异常的标志。
本研究发现,AICAR注射后1h游离性Beclin1蛋白含量显著下降(P<0.01),2h后基本恢复,可能Beclin1与其结合蛋白Vps34,PI3K等形成复合物,结合到自噬小体外膜,最终降解;Atg7在AICAR注射后1h也显著低于对照组(P<0.05),2h后基本恢复,尽管p62蛋白在AICAR注射后基本没有变化,但以上结果还是足以表明,AICAR诱导的AMPK激活可以显著提高骨骼肌安静状态下细胞自噬水平,具体机制为AMPK通过结合并磷酸化ULK1Ser317位点,提高ULK1酶活性,启动整个细胞自噬过程。
3.2 力竭运动诱导的AMPK激活对骨骼肌细胞自噬的影响
运动可以提高细胞自噬水平,这已经在心肌、肝脏、胰腺、脂肪和大脑皮质等多种组织和器官中得到证实[12,20]。研究人员发现,长期转轮运动可以上调小鼠骨骼肌LC3基因表达,提高自噬水平[26]。在胶原蛋白 VI敲除(Col6a1-/-)诱发的小鼠骨骼肌肌萎缩症中发现,自噬过程受损,Col6a1-/-鼠在长期转轮运动后,与Col6a1-/-鼠未运动组相比,LC3-II表达显著下降,LC3-II/LC3-I比值显著下降,小鼠骨骼肌纤维降解以及肌肉萎缩加重,有趣的是,正常鼠骨骼肌中,运动组LC3-II/LC3-I比值与对照组相比无显著性差异;为了排除长期运动引起的适应干扰,该研究组又分析了1h递增强度跑台运动对骨骼肌自噬的影响,发现LC3-I向LC3-II转化显著增加,组织化学分析发现自噬小体形成数目增多;而Col6a1-/-鼠在跑台运动后LC3脂化水平较低,与Col6a1-/-鼠未运动组相比无显著性差异[10]。以上实验提示,细胞自噬参与了骨骼肌对运动的应答。大量研究已经表明,运动激活AMPK,然而运动对ULK1影响尚未见报道。
本研究发现,力竭运动可显著提高骨骼肌AMPK活性,ULK1Ser317磷酸化水平以及AMPK与ULK1结合量,三者变化趋势基本一致,而且这种效应完全被AMPK抑制剂Compound C抑制,证明ULK1Ser317磷酸化是AMPK依赖性的。Kim等[16]发现小鼠以12.3m/min强度进行50min跑台运动后,LC3-II蛋白在恢复期3h、6h和12h显著下降,同时Atg7、Beclin1同步下降。本实验发现,在力竭运动后即刻,LC3-I含量下降非常明显(P<0.01),LC3-II含量显著增加,同时 LC3-II/LC3-1比值变大,表明自噬水平显著提高;3h时自噬仍处于较高水平,此后LC3-II持续下降,LC3-II/LC3-I比值也逐渐变小,表明LC3-II已经被降解,自噬水平下降,而LC3-I含量在6h时较运动后3h明显增加,LC3-II/LC3-I比值变化却并不明显,表明自噬水平没有差异,提示可能是运动诱导了LC3蛋白表达增多,使得LC3-I生成速度超过了LC3-I向LC3-II转化速度,超过了LC3-II降解速度,造成积累,随着时间推移,自噬水平的下降LC3-II和LC3-II/LC3-I比值进行性下降。同时游离性Atg7、Beclin1在运动后即刻下降显著,提示自噬过程被激活,其中Beclin1在6h时基本恢复,而Atg7在12h时才恢复。Compund C+力竭运动组小鼠 LC3-I、LC3-II、Beclin1、ATG7等变化趋势同力竭运动组,在运动后即刻均有显著性下降或提高,但下降或提高水平均不同程度弱于同一时刻取材的力竭组,提示除AMPK/ULK1途径外,运动训练诱导的细胞自噬激活还受其他途径调节,p62在Compound C注射后明显增多,再次表明运动诱导的骨骼肌细胞自噬过程受AMPK调控。
4 结论
1.AICAR诱导的AMPK激活显著提高小鼠骨骼肌AMPK与ULK1的结合,提高ULK1激酶活性,提高基础状态下骨骼肌自噬水平,表明AMPK是活体骨骼肌内细胞自噬的重要调节因子之一。
2.力竭运动可以通过AMPK/ULK1途径显著提高骨骼肌细胞自噬水平,但在Compound C诱导的AMPK活性抑制情况下,力竭运动仍能够显著提高骨骼肌细胞自噬水平,表明AMPK/ULK1并非是调节骨骼肌细胞自噬的唯一途径,运动应激可能通过其它途径提高自噬水平或ULK1活性还受其他磷酸化事件调控。
[1]钱帅伟,罗艳蕊,漆正堂,等.细胞自噬的分子学机制及运动训练的调控作用[J].体育科学,2012,32(1):64-70.
[2]张国华.不同运动骨骼肌AMPK的变化特点及对mTOR和下游信号的调控[D].北京:北京体育大学博士学位论文,2008.
[3]BARTH S,GLICK D,MACLEOD K F.Autophagy:assays and artifacts[J].J Pathol,2010,221(2):117-124.
[4]CAO Y,KLIONSKY D J.Physiological functions of Atg6/Beclin 1:a unique autophagy-related protein[J].Cell Res,2007,17(10):839-849.
[5]EGAN D F,SHACKELFORD D B,MILHAYLOVA M M,et al.Phosphorylation of ULK1(hATG1)by AMP-activated protein kinase connects energy sensing to mitophagy[J].Sci,2011,331(6016):456-461.
[6]FERNANDO P,BONEN A,HOFFMAN-GOETZ L.Predicting submaximal oxygen consumption during treadmill running in mice[J].Can J Phys Pharmacol,1993,71(10-11):854-857.
[7]GARBER K.Autophagy.Explaining exercise[J].Sci,2012,335(6066):281.
[8]GAIDHU M P,BIKOPOULOS G,CEDDIA R B.Chronic AICAR-induced AMP-kinase activation regulates adipocyte lipolysis in a time-dependent and fat depot-specific manner in rats[J].Am J Phys Cell Phys,2012,303(11):C1192-C1197.
[9]GE W,GUO R,REN J.AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol[J].Free Radic Biol Med,2011,51(9):1736-1748.
[10]GRUMATI P,COLETTOO L,SCHIAVINATO A,et al.Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles[J].Autophagy,2011,7(12):1415-1423.
[11]HE C,BASSIK M C,MORESI V,et al.Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis[J].Nature,2012,481(7382):511-515.
[12]HE C,SUMPTER R J,LEVINE B.Exercise induces autophagy in peripheral tissues and in the brain[J].Autophagy,2012,8(10):1548-1551.
[13]HOSOKAWA N,HARA T,KAIZUKA T,et al.Nutrient-dependent mTORC1association with the ULK1-Atg13-FIP200 complex required for autophagy[J].Mol Biol Cell,2009,20(7):1981-1991.
[14]JU J S,VARADHACHARY A S,MILLE S E,et al.Quantitation of“autophagic flux”in mature skeletal muscle[J].Autophagy,2010,6(7):929-935.
[15]KIM J,KUNDU M,VIOLLET B,et al.AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1[J].Nat Cell Biol,2011,13(2):132-141.
[16]KIM Y A,KIM YS,SONG W.Autophagic response to a single bout of moderate exercise in murine skeletal muscle[J].J Phys Biochem,2012,68(2):229-235.
[17]LEE J W,PARK S,TAKAHASH Y,et al.The association of AMPK with ULK1regulates autophagy[J].PLoS One,2010,5(11):e15394.
[18]MASIERO E,AGATEA L,MAMMUCARI C,et al.Autophagy is required to maintain muscle mass[J].Cell Metab,2009,10(6):507-515.
[19]MATSUIT,ISHIKAWA T,ITO H,et al.Brain glycogen supercompensation following exhaustive exercise[J].J Phys,2012,590(Pt 3):607-616.
[20]OGURA Y,IEMITSU M,NAITO H,et al.Single bout of running exercise changes LC3-II expression in rat cardiac muscle[J].Biochem Biophys Res Commun,2011,414(4):756-760.
[21]O'LEARY M F,VAINSHTEIN A,IQBAL S,et al.Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle[J].Am J Physiol Cell Physiol,2013,304(5):422-30.
[22]OTOMO C,METLAGEL Z,TAKAESU G,et al.Structure of the human ATG12~ATG5conjugate required for LC3lipidation in autophagy[J].Nat Struct Mol Biol,2013,20(1):59-66.
[23]SANCHEZ A M,CSIBI A,RAIBON A,et al.AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3aand interaction with Ulk1[J].J Cell Biochem,2012,113(2):695-710.
[24]SANDRI M.Autophagy in health and disease.3.Involvement of autophagy in muscle atrophy[J].Am J Phys Cell Phys,2010,298(6):C1291-C1297.
[25]WEBSTER I,FRIEDRICH S O,LOCHNER A,et al.AMP kinase activation and glut4translocation in isolated cardiomyocytes[J].Cardiovasc J Afr,2010,21(2):72-78.
[26]WOHLGEMUTH S E,SEO A Y,MARZETTI E,et al.Skeletal muscle autophagy and apoptosis during aging:effects of calorie restriction and life-long exercise[J].Exp Gerontol,2010,45(2):138-148.
